Материал: GMP_prefinal
-
Квалификация функционирования (Operational Qualification - OQ). Проводит-
ся проверка и оценка работоспособности объекта квалификации. Может проводиться как с использованием, так и без использования имитатора препарата.
-
Квалификация эксплуатации (Performance Qualification - PQ). Проводится про-
верка и оценка надежности и эффективности эксплуатационных параметров объекта ква-лификации. Проводится с использованием имитатора препарата или одной серии реально-го продукта.
Валидация аналитических методик
-
Каждая аналитическая и микробиологическая методика, которая используется для контроля качества сырья, полупродукта или готового продукта должна пройти валидацию. Это означает, что мы обязаны получить доказательства пригодности такой методики для контроля конкретного продукта и соответственно, гарантии получения достоверных ре-зультатов. В этом плане, требования GMP полностью совпадают с требованиями ИСО
17025.
Валидация очистки
-
Процедуры очистки оборудования должны также пройти валидацию до того, как мы приступим к производству препарата на этом оборудовании. Прежде всего, эта вали-дация направлена на получение гарантий возможности проведения качественной очистки после изготовления такого продукта. По сути, это минимизация риска перекрестного за-
51
грязнения при переходе на производство другого продукта на этом же оборудовании. Если на оборудовании останутся остатки предыдущего продукта, это не будет обнаружено – так как отсутствует аналитический контроль именно на наличие таких примесей.
Валидация асептических условий
-
При производстве стерильных лекарственных средств с использованием асептиче-ских технологий до начала самого технологического процесса необходимо подтвердить, что на всем протяжении процесса изготовления препарата (т.е. длительность процесса), в продукт не попадает ни один микроорганизм. Валидация асептических условий проводит-ся по сценарию имитации с помощью питательных сред.
Валидация технологического процесса
-
И непосредственно, валидация каждого из этапов технологического процесса про-водится на 3-х последовательных сериях с учетом «наихудшего случая». И, что очень важно, валидация технологического процесса проводится отдельно для каждого продукта
-
его заявляемого размера серии. Наихудший случай – это проведение процесса при таких условиях и обстоятельствах (для параметров процесса, режимов работы оборудования), которые имеют максимальные шансы вызвать отклонение процесса или несоответствие продукта по сравнению с идеальными условиями. Логика очень проста – если при таких условиях мы получаем качественный продукт, значит, гарантированно мы будем дости-гать качества внутри заданных диапазонов.
Виды валидации
-
Перспективная валидация
Проводится на вновь вводимом или реконструируемом производстве перед его пус-ком. При перспективной валидации обязательно проведение всех стадий квалификации (DQ, IQ, OQ, PQ) и валидации процессов и аналитических методов.
Выполняется при освоении новых технологий до серийного производства.
Состоит из 4-х этапов:
1 этап- включает в себя разработку планов проведения экспериментов по ста-диям процесса и, собственно проведение самих экспериментов для уточнения и опре-деления значений критических параметров стадий процесса.
2 этап - включает в себя окончательное согласование значений критических параметров стадий процесса и составление заключений.
3 этап - включает в себя разработку и утверждение программы валидации про-цесса и, собственно производство трех или больше серий продукта в пределах уста-новленных на 2 этапе.
4 этап- включает в себя составление и утверждение протокола о валидации процесса.
-
Сопутствующая (текущая) валидация
Проводится аналогично перспективной во время серийного производства, если оно не было валидировано ранее. При сопутствующей валидации обязательно проведение всех стадий квалификации (DQ, IQ, OQ, PQ) и валидации процессов и аналитических методов.
Проводится во время серийного производства, если оно не было валидировано ранее.
Применяется для процессов с историей стабильного качества.
Валидация проводится в объеме и в соответствии с 3 и 4 этапами перспектив-ной валидации с использованием ситуации наихудшего случая.
-
Ретроспективная валидация
52
Валидация процессов и аналитических методов проводится во время серийного про-изводства нестерильных лекарственных средств (если оно не было валидировано ранее) на основе анализа ранее полученных документально подтвержденных данных
Связана с исследованием накопленного опыта и проводится, если процедуры, оборудование и помещения не изменялись по крайней мере в течение одного года на момент проведения валидации или когда требование проводить валидацию произ-водства вводится впервые.
-
Ревалидация (Повторная валидация)
а) Проводится в плановом порядке в сроки, устанавливаемые предприятием в отчете о проведении валидации.
б) Проводится до возобновления производства в случаях изменения документации и/или условий производства, которые могут повлиять на качество полупродукта и готово-го продукта. Объем валидационных работ определяется предприятием исходя из внесен-ных изменений.
46. Определение стадий технологического процесса, требующих про-верки при валидации. Документирование валидации технологиче-
ского процесса.
Из методички АБ
Проводится валидаця критических стадий производственного процесса, оказываю-щих влияние на качество фармацевтических субстанций.
Критические параметры и (или) характеристики, как правило, следует определять на стадии разработки или на основании данных предварительного опыта работы; следует также определить диапазоны значений этих критических параметров и (или) характери-стик, необходимые для обеспечения воспроизводимости процесса. При этом необходимо:
-
определить критические характеристики ФС как продукции;
-
указать параметры процесса, которые могут влиять на критические показатели ка-чества ФС; установить диапазон значений каждого критического параметра процесса, ко-торый предполагается использовать при серийном производстве и контроле процесса.
Операции, которые считаются критическими для качества и чистоты ФС, подлежат валидации.
Документация по валидации
Для каждого процесса, подлежащего валидации, должен быть разработан протокол валидации. Этот протокол должен быть проверен и утвержден подразделением (подразде-лениями) качества и другими соответствующими подразделениями.
-
протоколе валидации должны быть определены критические стадии процесса и критерии приемлемости, а также вид проводимой валидации (например, ретроспективная, перспективная, сопутствующая) и количество производственных циклов.
Отчет о валидации должен содержать перекрестные ссылки на протокол валидации и обобщать полученные результаты, объяснять любые обнаруженные отклонения с соответ-ствующими выводами, включающими рекомендуемые изменения для исправления недо-статков.
Любые отклонения от протокола валидации должны быть оформлены документаль-но с соответствующим обоснованием.
53
47. Срок регистрации лекарственного средства закончился. Возможна его дальнейшая реализация? Если да, то, при каких условиях? От- вет обосновать
Согласно двум статьям из Федерального закона РФ №61-ФЗ “Об обращении лекар-ственных средств” делаем вывод что возможна продажа данного лекарственного средства
-
период проведения процедуры продления регистрации, если средство было ранее заре-гистрировано. Срок регистрации составляет 5 лет. А также согласно норме одной из ста-тей если срок действия регистрационного удостоверения на лекарственное средство за-кончился, его гражданский оборот на территории РФ не запрещен независимо от того, проводится ли в отношении данного лекарственного средства процедура подтверждения регистрации или нет.
Дополнительно:В соответствии с частью 1 статьи 13 Федерального закона РФ от 12.04.2010 г. N 61-ФЗ «Об обращении лекарственных средств» (в ред. от 06.12.2011) ле-карственные препараты вводятся в гражданский оборот на территории Российской Феде-рации, если они зарегистрированы соответствующим уполномоченным федеральным ор-ганом исполнительной власти. Таким образом, Закон указывает на запрещение ввода в
гражданский оборот незарегистрированного лекарственного средства, но не запрещает оборот лекарственных средств, которые введены в оборот на законном основании, но срок действия регистрационного удостоверения на которые закончился. Согласно части 7 ста-тьи 29 Закона «Об обращении лекарственных средств» в период проведения процедуры подтверждения государственной регистрации лекарственного препарата его гражданский оборот осуществляется на территории Российской Федерации.
Заметим, что приведенная выше формулировка части 7 статьи 29 не позволяет сде-лать однозначный вывод о том, можно ли в период проведения процедуры подтверждения государственной регистрации лекарственного средства вводить его в гражданский оборот.
-
то же время, в противном случае, норма части 7 статьи 29 Закона является избы-точной, поскольку согласно норме части 1 статьи 13 Закона если срок действия реги-
страционного удостоверения на лекарственное средство закончился, его граждан-ский оборот на территории РФ итак не запрещен, независимо от того, проводится ли
-
отношении данного лекарственного средства процедура подтверждения регистра-ции или нет.
Однако, трудно сказать, как контрольно-надзорные и правоохранительные органы будут трактовать указанные выше нормы Закона.
Тем не менее, по нашему мнению, если таможенные органы разрешили ввоз лекар-ственного препарата на территорию РФ в период его перерегистрации на основании под-тверждающего Письма Минздравсоцразвития РФ, то препарат введен в гражданский обо-рот на законных основаниях и его дальнейший оборот не будут являться нарушением дей-ствующего законодательства об обращении лекарственных средств.
+Время проведения клинического исследования лекарственного препарата не учитывается при исчислении срока его государственной регистрации.
48. Какого разрешительного документ фармацевтического предприя-тия нельзя получить без наличия утвержденного промышленного регламента? Технологический промышленный регламент на ле-
карственный препарат был утвержден в 2013 году. Потерял он свою силу? Ответ обосновать.
Промышленный регламент включает в себя перечень используемых фармацевтиче-ских субстанций и вспомогательных веществ с указанием количества каждого из них, данные об используемом оборудовании, описание технологического процесса и методов контроля на всех этапах производства лекарственных средств. Общие требования к струк-
54
туре и иные требования к содержанию промышленных регламентов установлены соответ-ствующими нормативными правовыми актами Российской Федерации.Без его утвержде-ния нельзя получить лицензию на фармацевтическую деятельность в России.
Срок действия ТР непосредственно зависит от того, на какой технологический про-цесс он был разработан:
1)Постоянный технологический регламент:срок действия — не более 10 лет. Каждые пять лет его действие необходимо подтверждать.
2)Временный технологический регламент:сроки утверждаются согласно действую-щим нормам по освоению производств и с расчетом времени, требуемого для разработки постоянного регламента. Допускается указывать срок действия регламента до одного года, при сроке освоения временного технологического регламента менее одного года. При отсутствии нормативов по освоению, действие документа прописывается лицом, его утверждающим.После завершения срока действия технологического временного регла-мента должен быть принят постоянный нормативно-технический документ.
3)Разовый технологический регламент:
срок действия устанавливается согласно датам осуществления опытных работ или срокам изготовления определенного объема товара. Поскольку, в соответствии с разовы-ми регламентами осуществляется наработка опытной партии товаров в течение несколь-ких лет, действие документа не должно превышать пять лет.
Срок действия Технологического регламента определяется законодательно, но, как правило, составляет 5 лет.. По истечении которых, если не произошло на производстве существенных изменений, то он продлевается еще на 5 лет. Если предприятие собирает-ся запустить выпуск новой продукции или ввести в строй новое оборудование, то тогда ТР разрабатывается на 2 года.Технологический регламент может перерабатываться досрочно
-
случаях, предусмотренных законодательством:
-
при введении новых законодательных актов по промышленной безопасно-
сти;
-
при принципиальных изменениях в технологии производства продукции;
-
если произошли аварии по причине того, что безопасные условия эксплуа-тации отражены в действующем ТР недостаточно.
49. Фармацевтическое предприятие производит выпуск новой продук-ции. Технолог-оператор на каждую производственную серию со-
ставлял протокол производства серии. Оценить действия техноло-га-оператора. Ответ обосновать.
На каждую изготовленную серию следует составлять и хранить протокол производ-ства серии. Он должен основываться на соответствующих частях утвержденных докумен-тов: производственной рецептуры и технологических инструкций.
Перед началом любого технологического процесса должно быть проверено и запро-токолировано, что оборудование и рабочая зона освобождены от предыдущей продукции, документов и материалов, не требующихся для планируемого процесса, и что оборудова-ние является чистым и готовым к эксплуатации.
-
ходе технологического процесса во время каждой проведенной действия инфор-мация о нем должна быть запротоколирована, по завершении операций протокол должен быть согласован, датирован и подписан лицом, ответственным за технологический про-цесс; протокол должен содержать следующую информацию:
- наименование продукции;
- дата и время начала и завершения технологического процесса, а также основных промежуточных стадий (операций);
- фамилия лица, ответственного за каждую стадию технологического процесса;
55
-
фамилию оператора различных важных этапов технологического процесса и при необходимости - лица, проверила каждую из этих операций (например, взвешивания);
номер серии и/или номер сертификата качества (аналитического паспорта
При необходимости должны быть даны ссылки на нормативные документы (ин-струкции) по охране труда и техники безопасности, требования которых необходимо со-блюдать при ведении описываемого технологического процесса.
Аналогично осуществляют инструкции относительно упаковки серии. Необходимо указать следующие данные:
-
теоретический выход - количество готовой продукции;
-
дата (время) начала и окончания всего процесса упаковки, который регламентиру-ется данным документом;
-
количество полученной нерасфасованной продукции и необходимые для упаковки материалы с указанием номера серии и/или сертификата качества (аналитического пас-порта).
Должны быть приведены данные о помещениях (код) и самое важное использован-ное оборудование (наименование, тип и номер). В инструкции должна содержаться указа-ние о необходимости идентификации упаковочной линии соответствующей идентифика-ционной этикеткой.
Экспертиза, согласование, утверждение рецептур и технологических инструкций и инструкций по упаковке такие же, как и для технологических и технических регламентов. Поэтому действия технолога-оператора считаются правомерными.
50. Составьте проект расположения производственных и вспомога-тельных помещений производства стерильных лекарственных форм с указанием вида деятельности в каждом из них: а) укажите классы чистоты помещений и дайте им характеристику, исходя из предъявляемых требований; б) укажите направление технологиче-
ских потоков в производственных помещениях.
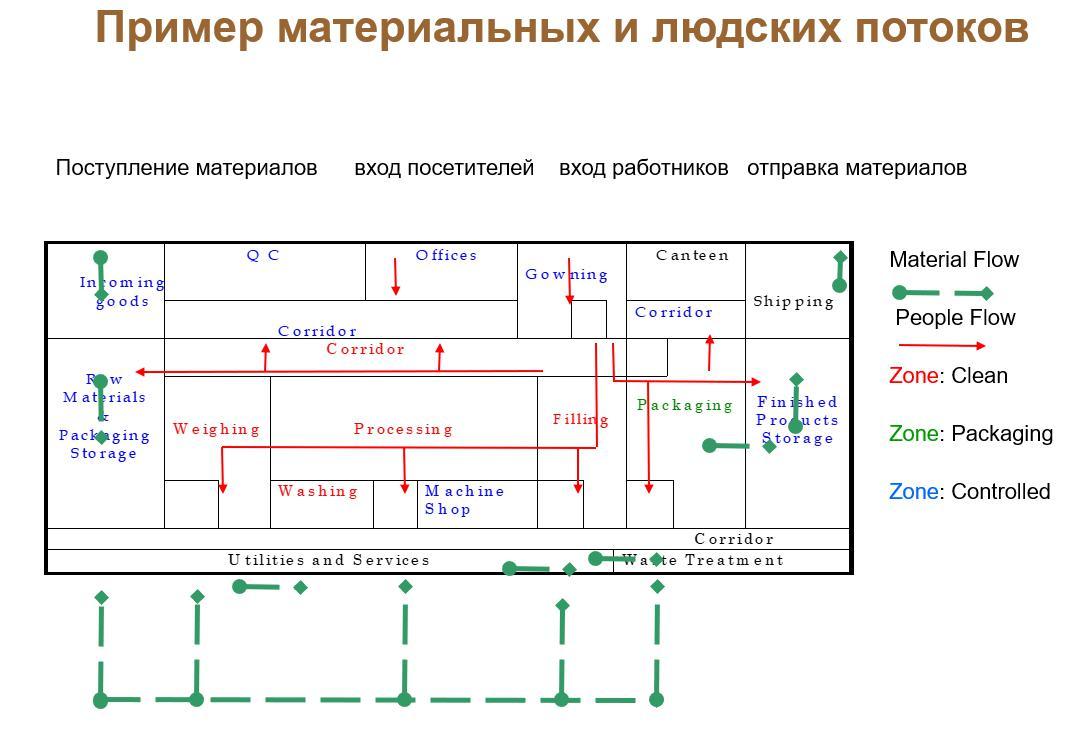
56
