Материал: GMP_prefinal
-
Кто является Уполномоченным лицом на фармацевтическом предприятии? Что входит в обязанности этого сотрудника? Кому могут быть, в случае необходимости, переданы его функции?
Уполномоченным лицом производителя лекарственных средств является его работник, аттестованный в установленном уполномоченным федеральным ор-ганом исполнительной власти порядке и имеющий стаж работы не менее чем пять лет в области производства и (или) контроля качества лекарственных средств, высшее образование соответственно по одной из специальностей и (или) одному из направлений подготовки: биология, биотехнология, ветерина-рия, клиническая медицина, радиационная, химическая и биологическая защита, фармация, фундаментальная медицина, химическая технология, химия. Соглас-но пункту 29 утвержденных Приказом Минпромторга РФ от 14.06.2013 г. N 916 «Правил надлежащей производственной практики» (в ред. от 18.12.2015) ква-лификация уполномоченного лица должна соответствовать требованиям, уста-новленным законодательством Российской Федерации. Уполномоченное лицо должно состоять в штате производителя. Его обязанности могут быть переданы только другому уполномоченному(ым) лицу(ам).
Руководящие работники – руководители производства, руководитель службы (отдела) контроля качества и уполномоченное лицо (лица) должны быть заняты на предприятии, как правило, полный рабочий день. Уполномоченное лицо (УЛ) играет важную роль в си-стеме качества фармацевтического производителя, увязывая основные элементы системы обеспечения качества, эффективности и безопасности лекарственных препаратов: реги-страцию продуктов, лицензирование производителей и правила GMP. В Европейском ре-гионе функции УЛ отражены не только в правилах GMP, но и в ряде важнейших отрасле-вых директив ЕС, в национальном законодательстве и подзаконных актах отдельных гос-ударств. Они находят поддержку в традициях и неписанных правилах, которыми руковод-ствуются надзорные органы, а в ряде европейских стран и в деятельности общественных профессиональных объединений.
Обязанности уполномоченных лиц
83
-
В отношении лекарственных средств, выпущенных в Российской Федерации, уполно-моченное лицо должно гарантировать, что каждая серия продукции была изготовлена и проверена в соответствии с установленными требованиями.
-
В отношении лекарственных средств, выпущенных за пределами Российской Федера-ции, уполномоченное лицо должно гарантировать, что импортируемая серия продукции прошла проверку в порядке, установленном в Российской Федерации.
-
До выдачи разрешения на выпуск лекарственных средств в сферу обращения уполно-моченное лицо должно документально подтвердить, что каждая серия продукции удовле-творяет требованиям, установленным при государственной регистрации.
Квалификация уполномоченного лица должна соответствовать установленным требовани-ям. Уполномоченное лицо должно входить в штат предприятия – производителя лекар-ственных средств. Его обязанности могут быть переданы только лицам, имеющим статус уполномоченного лица.
Ключевая функция УЛ
Ключевой обязанностью УЛ является решение вопроса о пригодности или непригодности каждой серии готовых лекарственных препаратов, законченной производством (на данном пред-приятии) к выпуску для продажи или бесплатного распределения. Выпуску подлежат только се-рии, все свойства продукции и условия производства которых, включая все виды контроля, соот-ветствуют всем относящимся к делу законам и правилам. На практике аналогичный подход часто применяется также к выпуску полупродуктов, подлежащих дальнейшей обработке на других пло-щадках.
-
наиболее общем случае пригодность серии означает, прежде всего, соответствие гото-вой продукции установленным спецификациям качества и соблюдение правил GMP. В отношении зарегистрированных лекарственных продуктов на первое место по важности выдвигается соблю-дение всех положений регистрационного досье.
-
этой связи следует еще раз подчеркнуть теснейшую связь правил GMP с системой реги-страции препаратов. Работать по GMP означает, прежде всего, обеспечить порядок, при котором по каждому выпускаемому лекарственному средству технология его производства, порядок внут-рипроизводственного контроля и выпуска готовых серий, а также спецификация качества готового лекарственного продукта соответствуют тому, что зафиксировано в регистрационном досье.
Как показывает практика, в целях эффективного выполнения ключевой функции выпуска серий УЛ также обязано:
• осуществлять постоянно верификацию системы качества предприятия и своевре-менно информировать руководство о наличии или угрозе возникновения существенных проблем
-
сфере качества; предлагать решения этих проблем;
-
быть в курсе всех изменений, касающихся соблюдения условий регистрации вы-пускаемых препаратов и правил GMP;
-
поддерживать хорошие деловые контакты с официальными инспекторами;
-
делегировать некоторые свои функции только хорошо подготовленным работни-
кам;

84
-
большинстве случаев УЛ несет ответственность за все аспекты качества в произ-водстве лекарственных продуктов на предприятии, иначе говоря, является верхов-ным контролером по качеству или арбитром в вопросах качества продукции
-
Кем, для чего и в какой форме создаются документы на фармацев-тическом предприятии? Как работник получает информацию о проведении необходимых операций/действий и сообщает об их ре-зультатах?
Документ - материальный объект, содержащий в зафиксированном виде инфор-мацию, оформленную установленным образом на определенном языке и носи-теле, имеющий в соответствии с действующим законодательством правовое значение.
Каждое предприятие должно разработать комплект документов, регламентиру-ющий производственную деятельность в соответствии с его профилем
-
Главной целью применяемой системы документации должно быть созда-ние, управление, контроль и регистрация всей деятельности, которая мо-жет непосредственно или опосредовано влиять на все аспекты качества лекарственных препаратов.
-
Существует два основных вида документации для выполнения требова-ний GMP и регистрации их соблюдения: инструкции (указания, требова-ния) и протоколы/отчеты.
Задачи системы документации:
-
обеспечить Уполномоченное лицо (лиц) информацией, необходимой для принятия решения о выдачи решения на реализацию серии продукции;
-
создать возможности для изучения несоответствий при выпуске продук-ции;
-
обеспечить информацией органы инспекции и регистрации для проведе-ния проверки условий производства и контроля качества продукции на предприятии;
-
обеспечить информацией органы инспекции для проведения проверки условий выпуска серии продукции, качество которой вызывает сомнения.
Ответственность за организацию и поддержание системы производствен-ной документации возлагается на:
-
Заместителя руководителя предприятия, курирующего вопросы каче-
ства (Директор по качеству) – обеспечивает общее руководство и кон-троль.
-
Начальника отдела обеспечения качества – согласование вновь созда-
ваемых документов, обеспечение своевременности пересмотра действу-ющих документов и поддержание жизнеспособности системы.
85
-
Контролера документов – текущая работа по соблюдению порядка об-ращения с документами.
-
Руководителей подразделений - актуализация и адекватность положе-ний о подразделении, должностных и рабочих инструкций
Необходимо обеспечить, чтобы документы были доступны всем исполни-телям, а устаревшая документация своевременно изымалась. Это означает:
-
Регулярную проверку документации (кем разработана, проверена, утвер-ждена, срок ее действия и соответствует ли она действующим НД).
-
Распределение документации, т.е. ее рассылка, учет и своевременное вне-сение изменений во все копии.
-
Устранение устаревшей версии документации.
Документы должны:
-
Утверждаться, подписываться и датироваться правомочными сотрудни-ками.
-
Иметь однозначное содержание.
-
Иметь ясный порядок и легко проверяться.
-
Регулярно анализироваться и обновляться.
-
Необходимо принять меры по недопущению использования устаревшей версии документов.
-
Все документы должны иметь срок и место хранения, быть доступными для использования, но иметь ограничения по пользователям.
-
Копии с документов должны быть четкими. Способ снятия копий с доку-ментов должен исключать возникновение ошибок.
-
Любые изменения в документах должны утверждаться в установленном на предприятии порядке.
-
При внесении в документы любых изменений небольшого объема, необ-ходимо указать причину, проставить дату внесения изменений и подпись внесшего изменения.
-
При необходимости внесения данных в документы (протоколы), следует это делать четко, разборчиво и так, чтобы внесенные сведения нельзя бы-ло стереть.
-
Полнота, аккуратность заполнения документов, а также точность внесе-ния и регистрации данных должны контролироваться.
-
Документы о совершении любых действий, позволяющие отслеживать все операции по производству продукции, должны храниться в установ-ленном месте в течение, как минимум, одного года со дня окончания сро-ка годности готовой продукции.
-
Данные могут регистрироваться электронным, фотографическим или дру-гим надежным способом.
86

Работник получает информацию о проведении необходимых операциях из до-кументов предприятия второго уровня, в частности из программ развития пред-приятия; производственных планов; должностных инструкций и СОПов (Стан-дартные операционные процедуры, рабочие инструкции и методики)
-
факте проведения необходимых операций работник сообщает руководству посредством написания отчёта (документ предприятия 3его уровня)
-
Охарактеризуйте параметры, которые необходимо подтвердить до начала технологического процесса при производстве стерильных лекарственных средств с использованием асептических техноло-гий. В каких условиях проводится валидация?
При производстве стерильных лекарственных средств с использованием асепти-ческих технологий до начала самого технологического процесса необходимо подтвердить, что на всем протяжении процесса изготовления препарата (т.е. длительность процесса), в продукт не попадает ни один микроорганизм. Вали-дация асептических условий проводится по сценарию имитации с помощью пи-тательных сред.
87

Анализ рисков в рамках испытания питательными средами– это не панацея от микробного загрязнения, но всего лишь набор инструментов, помогающий определить слабые места. Сам по себе он не принесет «исцеления» – он только «поставит диагноз» и в лучшем слу-чае «назначит лечение». В ходе оценки рисков определяются технологические операции, несущие риск нарушения стерильности продукта. Для каждой из этих операций иденти-фицируются источники (исходные компоненты, персонал, окружающая среда и т. д.), спо-собные эту стерильность нарушить. Исходя из обозначенных причин и степени критично-сти рис# ков, возможно выработать как способы их минимизации, так и методы оценки эффективности выбранных мероприятий.
88

66. Как определить риск для качества лекарственного средства? Ка-кие показатели качества контролировать и как часто? Предложите
алгоритм оценки рисков и разработки предупреждающих и кон-тролирующих действий.
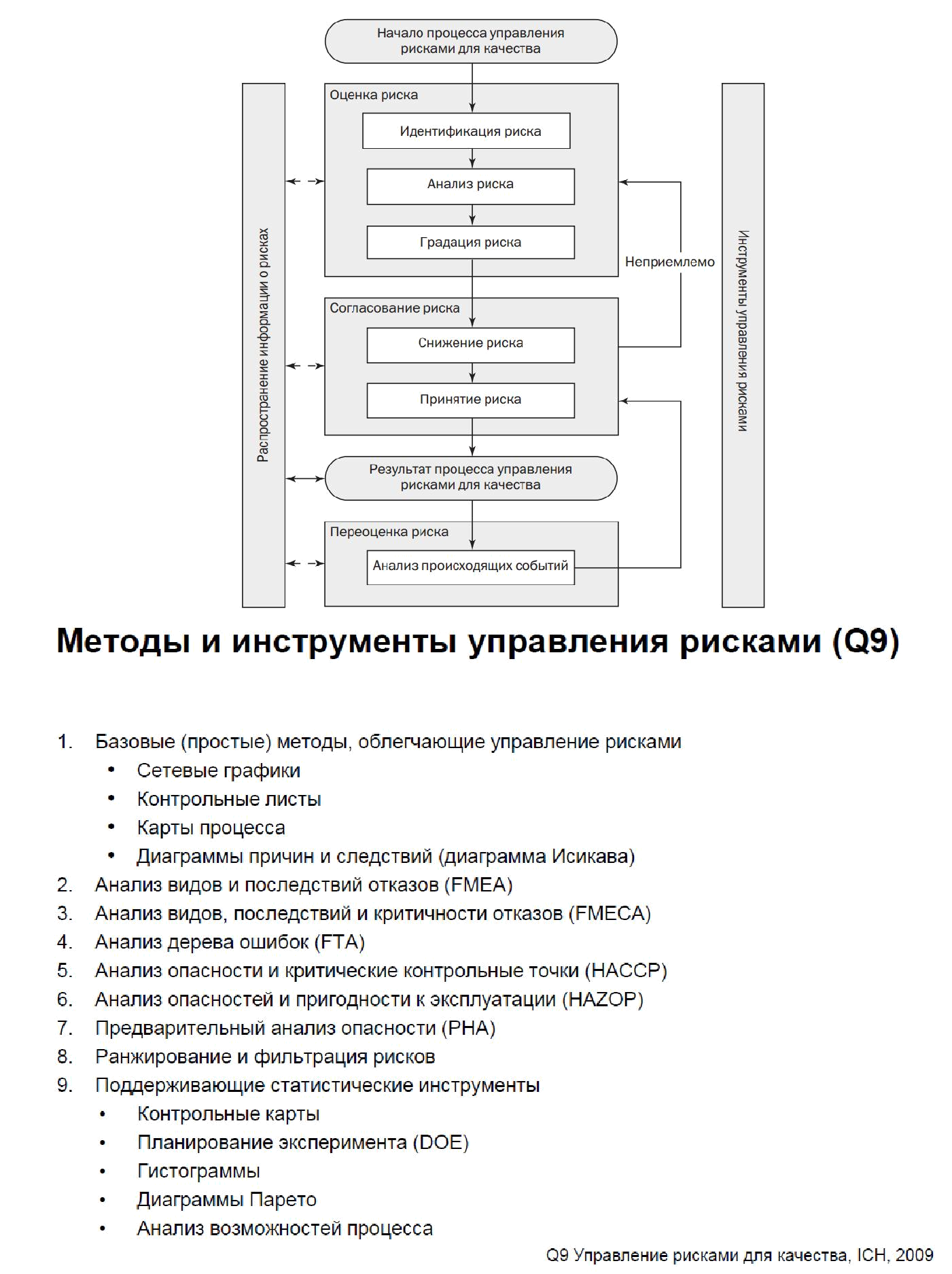
Схема управления рисками качества:
89
Причинно-следственный анализ
-
Структурированный метод идентификации возможных причин нежелательного со-бытия или проблемы.
-
Данный метод позволяет скомпоновать возможные причинные факторы в обоб-щенные категории так, чтобы можно было исследовать все возможные гипотезы.
90
Однако применение этого метода позволяет идентифицировать фактические при-чины.
-
Причины могут быть определены только на основе эмпирических данных или эм-пирическим путем. Информацию представляют в виде диаграммы "рыбьего скеле-та" (метод также называют диаграммой Исикавы) или иногда в виде древовидной схемы.
-
Наиболее целесообразно применять данный метод в самом начале анализа, что позволяет расширить диапазон представлений о возможных причинах, а затем сформулировать гипотезы, которые далее следует рассмотреть в соответствии с установленной процедурой.
-
Построение причинно-следственной диаграммы позволяет:
-
идентифицировать возможные первопричины и/или основные причины для опре-деленного следствия, проблемы или условия;
-
провести анализ в ситуации и найти взаимосвязь между взаимодействующими факторами, связанными с исследуемым процессом;
-
провести анализ существующих проблем для принятия корректирующих дей-ствий.
Основными этапами причинно-следственного анализа являются:
-
- установление следствия, которое необходимо проанализировать, и размещение его справа в соответствующем блоке диаграммы. Следствие может быть положи-тельным (цель) или отрицательным (проблема) в зависимости от обстоятельств;
-
- определение основных (главных) категорий причин и указание их в соответству-ющих блоках диаграммы "рыбьего скелета". При анализе систем обычно выделяют следующие категории причин: персонал, оборудование, рабочая среда, процессы и др. Категории определяют в соответствии с объектом исследования;
-
- указание возможных причин для каждой основной (главной) категории на ветвях
-
ответвлениях для описания взаимосвязей между ними;
-
-
- продолжение исследования путем итеративной постановки вопросов "почему?" или "что это вызвало?" для установления связей между причинами;