Материал: Совершенствование технологии получения адсорбентов из местных полиминеральных глин
В случае
цеолитов кислотными центрами являются отрицательно в заряженные
тетраэдры ![]() ,
возникающего в результате изоморфного замещения в тетраэдре
,
возникающего в результате изоморфного замещения в тетраэдре ![]() на
на ![]() , и катионы,
компенсирующие этот заряд, а также кислородные атомы алюмосиликатного каркаса.
Поэтому катионы цеолита, хотя и не являются каталитическим центром, своим
поляризующим действием могут влиять на активность кислотных центров цеолита
[62-69]. Эти взгляды целиком могут быть применены и в случае катализаторов
глинистого происхождения. В аспекте этого представляет интерес механизм
отравления каталитических центров цеолитов. Так, одно- и двухвалентные катионы
полностью компенсируют заряд тетраэдра
, и катионы,
компенсирующие этот заряд, а также кислородные атомы алюмосиликатного каркаса.
Поэтому катионы цеолита, хотя и не являются каталитическим центром, своим
поляризующим действием могут влиять на активность кислотных центров цеолита
[62-69]. Эти взгляды целиком могут быть применены и в случае катализаторов
глинистого происхождения. В аспекте этого представляет интерес механизм
отравления каталитических центров цеолитов. Так, одно- и двухвалентные катионы
полностью компенсируют заряд тетраэдра ![]() не зависимо от молекулярного
соотношения
не зависимо от молекулярного
соотношения ![]() в цеолите
типа А и Х. в случае же высококремнистых цеолитов типа У, у которых тетраэдра
в цеолите
типа А и Х. в случае же высококремнистых цеолитов типа У, у которых тетраэдра ![]() расположены
не через один, а через два тетраэдра
расположены
не через один, а через два тетраэдра ![]() , компенсирующие ионы, например,
катион
, компенсирующие ионы, например,
катион ![]() ,
располагаются асимметрично относительно соседних тетраэдров [62]. Благодаря
этому имеет место катиона по
,
располагаются асимметрично относительно соседних тетраэдров [62]. Благодаря
этому имеет место катиона по ![]() , в результате чего заряд одного
, в результате чего заряд одного ![]() будет компенсирован,
а другого - не компенсированным по схеме:
будет компенсирован,
а другого - не компенсированным по схеме:
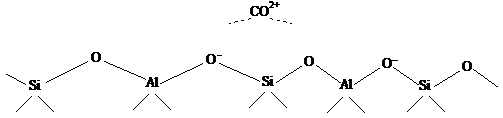
Отрицательный заряд ![]() и служит
активным центром алюмосиликата, а из газообразных
и служит
активным центром алюмосиликата, а из газообразных ![]() и веществ
содержащих аминогруппы
и веществ
содержащих аминогруппы ![]() пиридин и
т.п.
пиридин и
т.п.
Содержание воды в катализаторах
крекинга имеет некоторое значение при установлении связи между кислотностью и
химической природой кислотных центров катализатора. Ясно, что после прокалывания,
например, при 800º,
на
поверхности катализатора не остается адсорбционно связанной воды. Однако,
водород присутствует в виде гидроксильных групп, связанных с атомами кремния
или алюминия. Вероятно, водород находится у атома кремния, так как энергия
связи ![]() (1541,25
кДж/моль) больше против энергии связи
(1541,25
кДж/моль) больше против энергии связи ![]() (329,0 кДж/моль). Поэтому при
прокаливании водород удаляется из атома алюминия. Возникающая при этом
вакантность (электронный дефицит) компенсируется за счет миграции водорода от
силанальных групп. И этот водород является кислотным центром бренстедовского
(протонного) типа алюмосиликатного катализатора и у целотов [65, 66].
Установлено, что катализатор с большой величиной протонной кислотности является
одновременно хорошим катализатором в реакциях крекинга углеводородов [67-69].
(329,0 кДж/моль). Поэтому при
прокаливании водород удаляется из атома алюминия. Возникающая при этом
вакантность (электронный дефицит) компенсируется за счет миграции водорода от
силанальных групп. И этот водород является кислотным центром бренстедовского
(протонного) типа алюмосиликатного катализатора и у целотов [65, 66].
Установлено, что катализатор с большой величиной протонной кислотности является
одновременно хорошим катализатором в реакциях крекинга углеводородов [67-69].
Результаты работа Т. Г. Мелликкена и
др. [70, 71] позволяют предположить, что большинство албмосиликатных
катализаторов представляют собой смесь частиц двуокиси кремния и окиси алюминия
с ионами кремния и алюминия в решетке, совместно владеющим ионами кислорода.
Поэтому в смешанной окисной структуре будут проявляться химические свойства
окиси алюминия в ее различных кристаллических формах, тогда как габитусы кристаллов
двуокиси кремния будут играть в определении характера катализатора лишь
второстепенную роль. Бемит, байерит и гидраргиллит представляют собой
окисно-алюминиевые структуры с основными свойствами, и в их кристаллах алюминий
имеет координационное число шесть, будучи связанным с шестью атомами кислорода.
Так как радиус иона алюминия сравнительно невелик (около 0,5![]() ), то он
может перейти из состояния, характеризующегося координационным числом 4, в
состояние с координационным числом 5, вступая в коорданиционную связь с
четырьмя, либо с шестью плотно упакованными ионами кислорода. Кислотно-основные
свойства окиси алюминия определяются тем, какое координационное число имеют
ионы алюминия, образующие кристаллическую структуру. Если алюминий имеет
координационное число четыре, он ведет себя как кислота. Соли такой кислоты
имеют кольцевую структуру, состоящую из шести алюмокислородных тетраэдров с
катионом внутри этого кольца. В то же время двуокись кремния в кварце, тридимите
и кристобалите всегда имеет координационное число, равное четырем, причем
существование этих различных кристаллических форм обусловлено различным
расположением кремнекислородных тетраэдров в смесях окислов или в
алюмосиликатах. Стабильная тетраэдрическая структура кремния усилена
соответствующей координацией ионов кислорода вокруг иона алюминия, причем
габитус кристалла стабилизируется такими катионами, как ионы натрия или
алюминия. Так как кристобаллит имеет структуру, аналогичну структуре алюмината
калия, явно выраженные кольца кристобаллита образуются в тех точках
соприкосновения, где ионы кислорода делятся между двуокисью кремния и окисью
алюминия.
), то он
может перейти из состояния, характеризующегося координационным числом 4, в
состояние с координационным числом 5, вступая в коорданиционную связь с
четырьмя, либо с шестью плотно упакованными ионами кислорода. Кислотно-основные
свойства окиси алюминия определяются тем, какое координационное число имеют
ионы алюминия, образующие кристаллическую структуру. Если алюминий имеет
координационное число четыре, он ведет себя как кислота. Соли такой кислоты
имеют кольцевую структуру, состоящую из шести алюмокислородных тетраэдров с
катионом внутри этого кольца. В то же время двуокись кремния в кварце, тридимите
и кристобалите всегда имеет координационное число, равное четырем, причем
существование этих различных кристаллических форм обусловлено различным
расположением кремнекислородных тетраэдров в смесях окислов или в
алюмосиликатах. Стабильная тетраэдрическая структура кремния усилена
соответствующей координацией ионов кислорода вокруг иона алюминия, причем
габитус кристалла стабилизируется такими катионами, как ионы натрия или
алюминия. Так как кристобаллит имеет структуру, аналогичну структуре алюмината
калия, явно выраженные кольца кристобаллита образуются в тех точках
соприкосновения, где ионы кислорода делятся между двуокисью кремния и окисью
алюминия.
Некоторые авторы предполагают, что в
прокаленном алюмосиликатном катализаторе присутствует кислотный комплекс типа ![]() , обладающий
способностью диссоцировать с образованием протонов [72]. Однако, согласно
данным работы А. Г. Облада и др. [71]. Эта структура отвечает ангидриду
метаалюминиевой кислоты
, обладающий
способностью диссоцировать с образованием протонов [72]. Однако, согласно
данным работы А. Г. Облада и др. [71]. Эта структура отвечает ангидриду
метаалюминиевой кислоты ![]() , она не
способна диссоциировать, давая протоны, а потому ее следует считать кислотой
Льюиса, а не кислотой бренстеда.
, она не
способна диссоциировать, давая протоны, а потому ее следует считать кислотой
Льюиса, а не кислотой бренстеда.
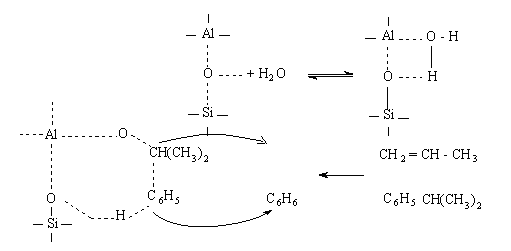
Согласно создавшемуся представлению об активном центре катализатора, углеводород, находящийся в газовой фазе, отдает электрон льюисовскому кислотному центру, при этом на поверхности катализатора образуется карбониевый ион, что стабилизирует кислотный центр катализатора. Стремление алюминия вернуться в состояние, характеризующееся координационным числом 6, создает движущую силу десорбции. Однако, для перемещения иона водорода, который также образуется в процессе крекинга, необходимо одновременно присутствие и сосуществование на двойном центре кислоты Льюиса и кислоты бренстеда [63]. Транспорт протонов может происходить и без участия кислоты бренстеда.
Ряд авторов, учитывая возможную
координационную связь у атома алюминия в алюмосиликатах, активным центром в них
считают атом алюминия, имеющий координационное число 3. при взаимодействии
молекул, образующих карбонион с алюмосиликатом, в том числе у цеолитового
крекинга, координационное число у атома алюминия становится равным 4 [28, 36,
70, 71] по схеме:
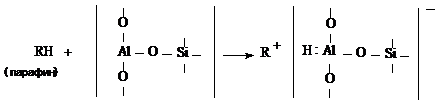
При этом допускают, что обменные свойства алюмосиликатов обусловлены именно таким атомом алюминия, ибо он, гидротируясь в водной среде, способствует образованию алюмосиликатного комплекса, при котором выделяется ион водорода [28, 52].
Таким образом, для алюмосиликатов
характерно наличие протонных и апротонных форм кислотности. Причем, их
содержание, а также соотношение могут регулироваться активацией алюмосиликатов.
Так, при обработке алюмосиликатов (например, глин) в начале, когда концентрация
активатора низка, происходит эквивалентный обмен поглощенных катионов твердой
фазой на катион дисперсионной среды. При этом получается Н-форма глины [4, 21,
31] . Однако, эта форма глины не устойчива во времени и переходит в ![]() -форму (или
-форму (или ![]() -форма
переходит в Н-форму) по параболическим законам [73]. Поэтому при обработке глин
разбавленными растворами солей алюминия или минеральных кислот образуются
одновременно Н и
-форма
переходит в Н-форму) по параболическим законам [73]. Поэтому при обработке глин
разбавленными растворами солей алюминия или минеральных кислот образуются
одновременно Н и ![]() -формы
глины. При обработке глины раствором кислоты повышенной концентрации происходит
разрушение кристаллической структуры глины и при этом происходит образование
алюмосиликата с повышенным содержанием
-формы
глины. При обработке глины раствором кислоты повышенной концентрации происходит
разрушение кристаллической структуры глины и при этом происходит образование
алюмосиликата с повышенным содержанием ![]() , обладающего кислотными свойствами,
о причине появления которых сказано выше.
, обладающего кислотными свойствами,
о причине появления которых сказано выше.
В работе [70] авторы допускают, что
при кислотной обработке глины, чтобы занаять место иона алюминия в глине, ион
водорода должен ассоциироваться с молекулами воды, тогда как ион![]() анимает
место большего катиона. Это предположение является стратегическим критерием
расположения активного центра с ионами алюминия, находящимися в
непосредственной близости от тетраэдрического кремнезема, и алюминий легко
меняет координационное число от 6 до 4. Если в процессе катализа при
взаимодействии кислого иона алюминия с молекулой углеводорода на поверхности
катализатора образуется ион углеводорода, то возникает вопрос, каким образом
ион углеводорода разлагается и как происходит десорбция продукта. Можно
считать, что тенденция иона алюминия вновь принимать шестерную координацию
является движущей силой десорбции. Следовательно, каталитическая активность
алюмосиликата зависит от легкости, с которой ион алюминия меняет свое
координационное число от 4 до 6. эти предположения подтверждаются работами ряда
авторов [72,].
анимает
место большего катиона. Это предположение является стратегическим критерием
расположения активного центра с ионами алюминия, находящимися в
непосредственной близости от тетраэдрического кремнезема, и алюминий легко
меняет координационное число от 6 до 4. Если в процессе катализа при
взаимодействии кислого иона алюминия с молекулой углеводорода на поверхности
катализатора образуется ион углеводорода, то возникает вопрос, каким образом
ион углеводорода разлагается и как происходит десорбция продукта. Можно
считать, что тенденция иона алюминия вновь принимать шестерную координацию
является движущей силой десорбции. Следовательно, каталитическая активность
алюмосиликата зависит от легкости, с которой ион алюминия меняет свое
координационное число от 4 до 6. эти предположения подтверждаются работами ряда
авторов [72,].
Известна проматирующая роль воды в
каталитическом процессе, если в качестве катализатора используется алюмосиликат
[61,71].
Установлено, что полностью обезвоженный алюмосиликат не активен в реакции
крекинга кумола [71]. Однако, если этот образец снова
гидратировать, его первоначальная активность восстанавливается. Проматирующее
действие небольших количеств воды в реакциях углеводородов объясняется тем, что
вода вызывает десорбцию карбоний-иона, сдвигая равновесие в сторону образования
новых ионов. Большое количество воды приводит к конкуренции за активным центром
между углеводородом и водой, и адсорбируются из них более полярные, т.е. воды,
что приводит к понижению активности катализатора. Однако,
одновременное существование донора протонов (кислоты)и акцептора протонов (основания)
в алюмосиликатах делает их способными для расщепления углеводородов [71].
Вода в алюмосиликатах, повидимому, одновременно служит акцептором и донором
протона, согласно схеме:
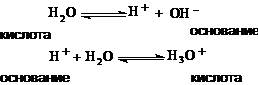
повышение каталитической активности прокаленных, затем охлажденных до температуры крекинга природных промышленных алюмосиликатов объясняется неравномерным распределением ОН-групп и высокой подвижностью протона вновь адсорбированной влаги и их миграцией на поверхности [70]. Сказанное подтверждается еще и тем, что при постоянном суммарном значении общей кислотности алюмосиликатного катализатора с содержанием определенного количества воды протонная кислотность катализатора имеет наибольшее значение при низких, а апротонная - при высоких температурах [35, 80].
Есть указание о том, что активность
катализатора уменьшается с ростом радиуса катиона, адсорбированного
алюмосиликатом [81]. Предполагают, что катализ в некоторых алюмосиликатах, в
частности на цеолитах, протекает не на протонных, а не катионных центрах,
имеющих электротстатическое поле сильнее, чем на протонных центрах. Молекула
углеводорода на них поляризуется (![]() ) или даже отщепляется с
образованием иона карбония.
) или даже отщепляется с
образованием иона карбония.
При оценке активности природных
алюмосиликатов (глин и др.) при крекинге кумола или газойливой фракции исходят
из суммарной величины обменных ионов ![]() и
и ![]() и отрицательного заряда решетки.
Замечена симбатность между изменением общеобменной кислотности алюмосилиткатов
при их активации и их каталитической активностью в реакциях крекинга [10, 18,
36, и др.].
и отрицательного заряда решетки.
Замечена симбатность между изменением общеобменной кислотности алюмосилиткатов
при их активации и их каталитической активностью в реакциях крекинга [10, 18,
36, и др.].
Таким образом, у алюмосиликатных
катализаторов имеются каталитические центры, проявление которых, в основном,
зависит от структуры алюмосиликата, наличия воды в нем и от процесса катализа.
Структура природных алюмосиликатов, количество воды в них и связанные с этим
свойства природных алюмосиликатов регулируются воздействием на них физическим и
химическим способами активации. При этом, в зависимости от условий активации,
образуются катализаторы, свойства которых устанавливаются пока опытным путем.
Можно с уверенностью сказать, что при воздействии на природный алюмосиликат
каким-либо методом активации в определенном участке его структуры происходит
изменение, благодаря чему возникает напряженность в структуре в целом, а в
системе появляется сила, направленная на уменьшение напряжения, возникшего в
результате внешнего воздействия на алюмосиликат. Однако, отсутствие прямого
измерения силы возникшего напряжения делает это положение гипотетическим.
Несмотря на это, увеличение адсорбционной и каталитической активности природных
алюмосиликатов при их термо-, термокислотнощелочно, и наоборот, активации
указывает на избыточность внутренней энергии, для компенсации возникающей
внешнего напряжения. Такое предположение согласуется с мнением Д.И.Менделеева о
внутренней подвижности составляющих системы при ее встрече с внешними
молекулами. Другими словами, адсорбенты и катализаторы являются более эффективными,
если в нем созданы благоприятные условия для внутреннего движения. Таким
образом, «катализатор - это приведенное в неустойчивое состояние вещество,
химически возбуждающее субстрат и направляющее химическую реакцию».
1.3
Физико-химические свойства Навбахорского бентонита
Как уже упоминалось, месторождение бентонитовых глин Навбахор было открыто в 1998 году [45]. Разведочные работы по нему завершены, запасы утверждены ГКЗ РУз и оно подготовлено к промышленному освоению.
Вещественный состав бентонитовых глин изучен авторами [46] комплексными лабораторными методами (химический, термический, электронно-микроскопический и рентгено-структурный анализы), определена общая обменная емкость поглощенных оснований, соотношение обменных катионов и изучены физико-химические свойства. По результатам лабораторных исследований в разрезе продуктивной толщи месторождения выделены щелочные и щелочно-земельные разновидности глин, которые сильно отличаются по физико-химическим свойствам друг от друга. Бентонитовое число (набухаемость) щелочных бентонитов в пробах колебалась от 42 до 86, в среднем 79мл. Коллоидальность изменялась от 45 до 90,6, в среднем 80,5%. Эти показатели в щелочноземельных бентонитах значительно ниже, чем в щелочных и составляют в среднем 41 мл и 51%, соответственно.
Объектами нашего исследования были щелочные
бентониты. Химический состав образца приведены ниже.
Химический состав (масс, %)
|
SiO2 |
AI2O3 |
Fe2O3 |
Na2O |
K2O |
MgO |
CaO |
P2O5 |
TiO2 |
FeO |
Ппп |
|
54,20 |
11,78 |
9,29 |
3,36 |
2,53 |
1,77 |
1,34 |
0,15 |
1,37 |
- |
14,21 |
Как видно из приведенных данных основным оксидом в минерале является оксид кремния ( 54,20 % масс.), содержание красящих оксидов тоже значительно: Fe2O3 - 9,29 и TiO2 - 1,37; оксида алюминия - 11,78% масс. На основании проведенных исследований можно сказать, что исследуемый образец сложен монтмориллонитом.
Адсорбционные характеристики, полученные из изотерм адсорбции паров воды и бензола методом БЭТ, (заимствовано из [59] показывают, что удельная поверхность по воде (237м2/г) больше, чем по бензолу (67м2/г)). Это обусловлено различием взаимодействий глин с полярными и неполярными молекулами адсорбата.
Изотермы адсорбции паров воды нового адсорбента измеренные, при 293К имеют S-образную форму. В работе [60] сорбционные характеристики, определенные с помощью метода БЭТ имеют следующие характеристики: ёмкость монослоя (аn) - 4,81моль/кг; удельная поверхность (S) - 319м2/г; пределы адсорбции (as) - 20,2 моль/кг; предельный адсорбционный объем (Vc) - 0,36 · 103 м3/кг.
Для определения термостойкости адсорбента и выяснения его поведения в процессе термообработки использован термический анализ (ДТА).
В работе [60] проведены термический анализ, рентгенографические и электронно-микроскопические (РЭМ) исследования [61,62] образцов Навбахорского бентонита.