Материал: Шмид Р. Наглядная биотехнология и генетическая инженерия
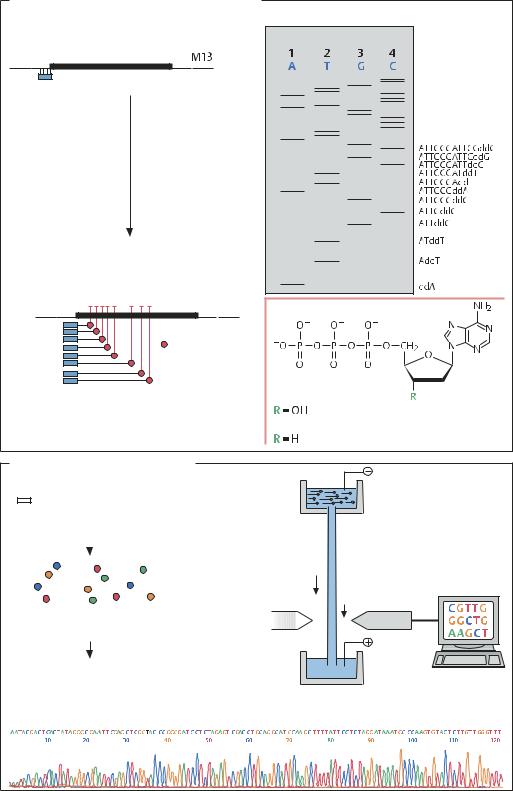










 Детектор
Детектор
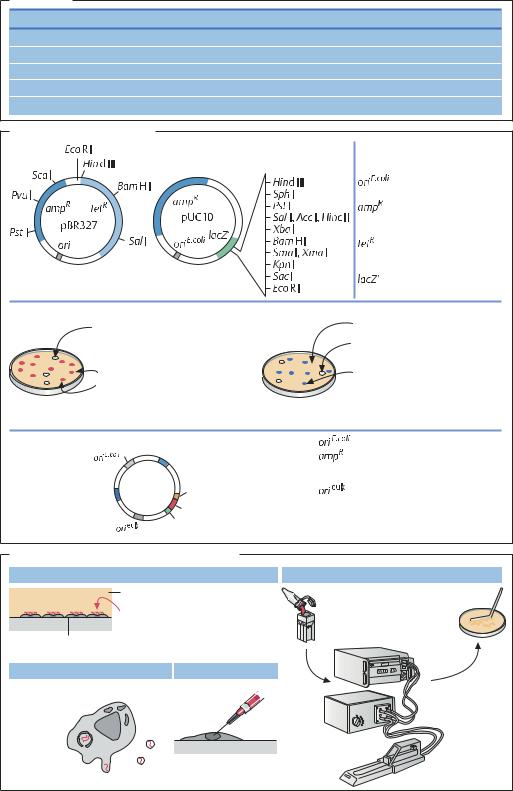
Плазмиды |
|
|
|
|
|
|
Пример |
Размер, т. п. н. |
Источник |
|
|
F (фактор фертильности) |
F-плазмида |
95 |
Escherichia coli |
|
|
R (фактор резистентности) |
RP4 |
54 |
Pseudomonas sp. |
|
|
T (колицин) |
ColE1 |
6,4 |
Escherichia coli |
|
|
Плазмида деградации |
TOL |
117 |
Pseudomonas putida |
|
|
Плазмида вирулентности |
Ti-плазмида |
213 |
Agrobacterium tumefaciens |
|
|
Вектор для клонирования |
MCS (multiple cloning site) – |
Два вектора для клонирования |
|
||
|
|
||||
|
полилинкерный участок |
в E. coli |
|
|
|
|
с уникальными сайтами для |
|
|
|
|
|
расщепления рестриктазами |
|
|
|
|
|
|
|
|
точка начала |
|
|
|
|
|
репликации в E. coli |
|
|
|
|
|
селективный маркер 1 |
|
|
|
|
|
(устойчивость |
|
|
|
|
|
к ампициллину) |
|
|
|
|
|
селективный маркер 2 |
|
|
|
|
|
(устойчивость |
|
|
|
|
|
к тетрациклину) |
|
|
|
|
|
фрагмент гена |
|
|
|
|
|
β-галактозидазы |
|
Скрининг по устойчивости к антибиотику |
«Бело-голубой» скрининг |
|
|
||
ampR tetS – рекомбинантный клон: |
|
Aгар + X-Gal + IPTG + ампициллин |
|
||
ген встроен по сайтам BamHI |
|
Белые колонии – рекомбинантные |
|
||
и Sal I – устойчивость |
|
|
|||
к тетрациклину утеряна |
|
клоны (lacZ' нарушен) |
|
||
ampR tetR – нерекомбинантный |
|
Голубые колонии – |
|
||
|
нерекомбинантные клоны |
|
|||
клон: устойчив к ампициллину |
|
|
|||
|
(lacZ' не нарушен) |
|
|||
и тетрациклину |
|
|
|
||
|
X-Gal – хромогенный субстрат β-галактозидазы |
|
|||
Агар, содержащий ампициллин и тетрациклин |
|
||||
IPTG – индуктор промотора β-галактозидазы |
|
||||
Челночный вектор для экспрессии в эукариотических клетках |
точка начала репликации в E. coli |
|
|||
Устойчивость |
|
|
|||
|
селективный маркер |
|
|||
|
эукариотических клеток |
|
|||
|
Терминатор |
|
(устойчивость E. coli |
|
|
|
|
к ампициллину) |
|
||
ampR (устойчивость |
транскрипции (сигнал |
|
|||
точка начала репликации |
|
||||
полиаденилирования) |
|
||||
E. coli к ампициллину) |
Полилинкер |
|
для эукариот (например, |
|
|
|
|
плазмиды размером 2 мкм |
|
||
|
Эукариотический |
|
в S. cerevisiae или вируса SV-40 |
|
|
|
промотор транскрипции |
в животных клетках) |
|
||
Небиологические методы трансформации |
|
|
|
|
|
Эндоцитоз |
|
|
Электропорация |
|
|
Раствор фосфата кальция |
Компетентные |
Распределение |
|
||
|
|
клетки |
|
по агару, |
|
ДНК оседает на клеточной |
и чужеродная |
|
|||
содер- |
|
||||
поверхности |
ДНК |
|
|
||
|
жащему |
|
|||
Монослой животных клеток |
|
|
|
антибиотик |
|
Липофекция |
Микроинъекция |
|
|
|
|
Перенос ДНК в клеточное ядро |
Раствор ДНК |
|
|
|
|
|
|
|
|
|
|
|
Клеточное |
|
|
|
|
Липосома, |
ядро |
|
|
|
|
|
|
|
|
|
|
слившаяся |
|
|
|
|
|
с клеткой |
|
|
|
|
|
(ДНК обозначена |
|
|
|
10 000 В/м2 |
|
красным цветом) |
|
|
|
в течение 10 мс |
243 |
|
|
|
|
||
|
|
|
|
|
|