Материал: GMP_prefinal
• Государственная регистрация лекарственных препаратов для медицинского приме-нения осуществляется по результатам экспертизы лекарственных средств и этической экспертизы возможности проведения клинического исследования лекарственного препа-рата для медицинского применения (далее - этическая экспертиза).
• Государственная
регистрация лекарственных препаратов
для ветеринарного при-менения
осуществляется по результатам экспертизы
лекарственных средств для ветери-нарного
применения.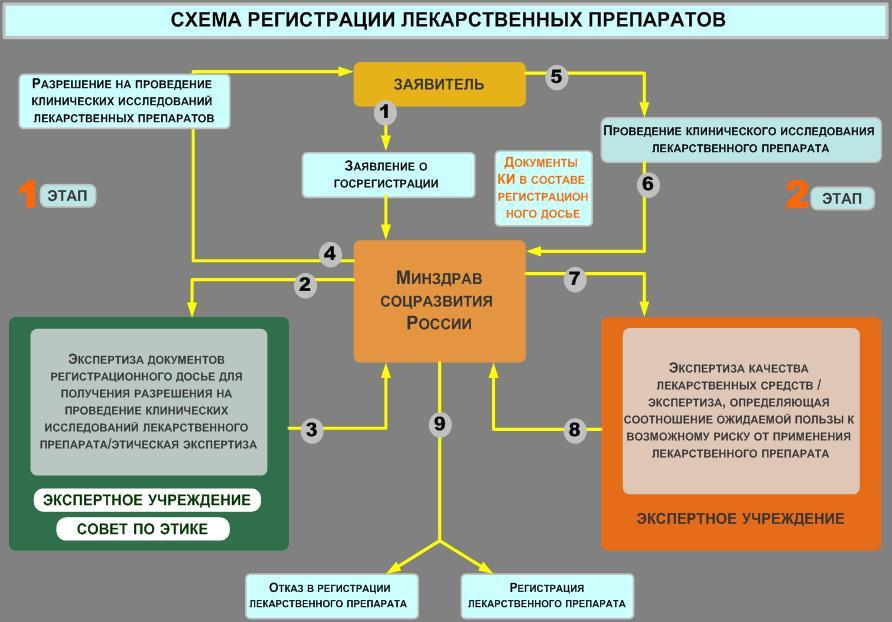
25. Охарактеризуйте правила лабораторной практики (glp).
-
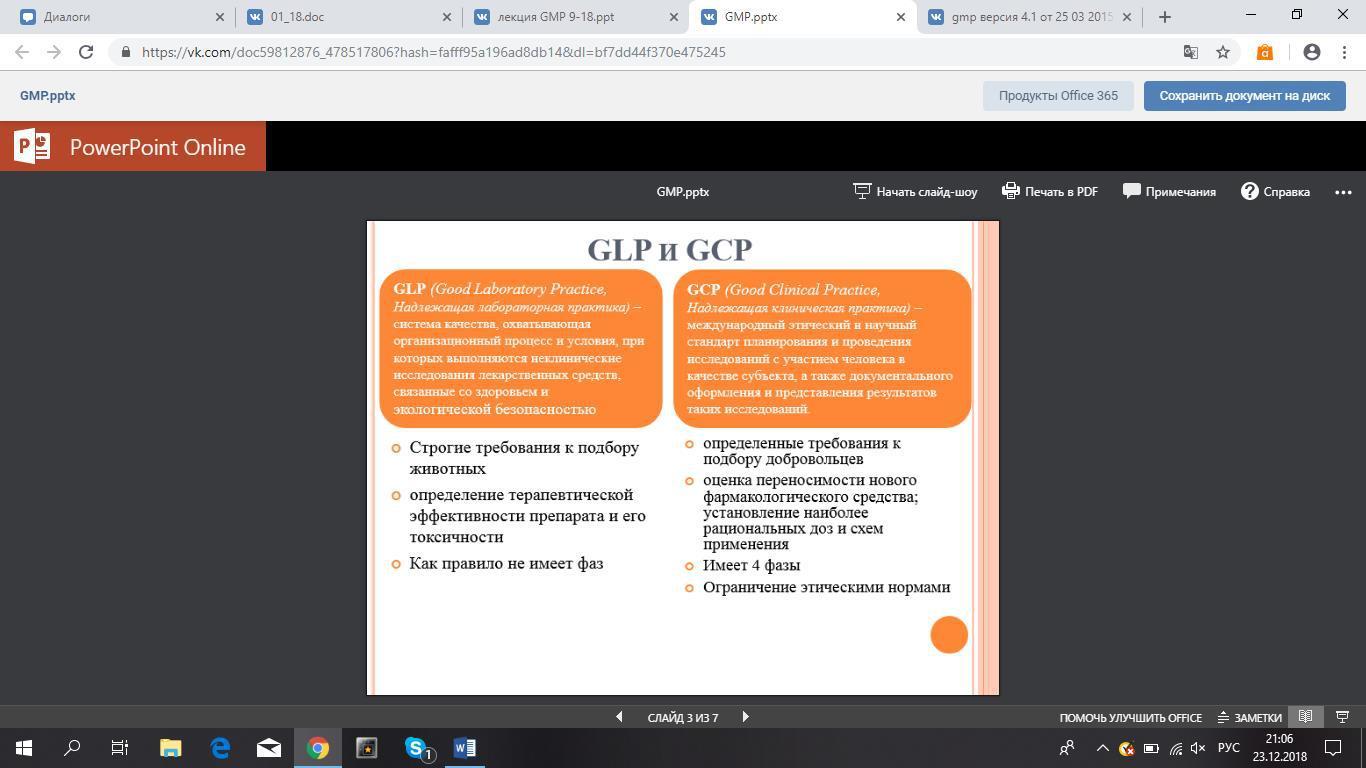
Правила лабораторной практики (GLР) распространяются на работу фармаколо-гических, токсикологических и других лабораторий биологического профиля и имеют це-лью обеспечение приемлемости научных исследований на этапе доклинического (точнее, неклинического) изучения новых препаратов. Под приемлемостью в данном случае пони-мается, с одной стороны, надежность и доказательность, с другой – соблюдение принци-пов гуманного обращения с лабораторными животными.
-
В ряде стран аналогичные правила распространяются на изучение токсичности хи-мических соединений, используемых в производстве потребительских (нелекарственных) товаров и продукции промышленного назначения (например, красителей, клеев, отделоч-ных материалов, продуктов агрохимии и др.), и их потенциальной опасности для окружа-ющей среды.
26. Дайте развернутую характеристику правил клинической практики (gсp).
Надлежащая клиническая практика (Good Clinical Practice; GCP) представляет собой международный этический и научный стандарт планирования и проведения исследований
-
участием человека в качестве субъекта, а также документального оформления и пред-ставления результатов таких исследований.
-
Под правилами «Надлежащей (или качественной) клинической практики» (GСР) понимается стандарт проведения клинического испытания, разработанный для того, что-бы предотвратить ошибки и подлог в процессе испытания лекарственного препарата и за-щитить права субъекта испытания.
-
Принципы GСР охватывают планирование, организацию, мониторинг, аудит, ана-лизы, отчетность и документацию клинического испытания, а также гарантируют, что это исследование научно и этически обосновано.
-
Правила GСР являются логическим продолжением GМР в области клинических исследований. Главное – стандартизовать подготовку исследования, сбор данных, их про-верку и анализ, ведение документации.
-
Правила определяют обязанности фармацевтической промышленности (спонсоров исследований), клинических исследователей и тех, кто контролирует ход исследований. В назначение этих правил также включены указания о порядке получения согласия пациен-тов или здоровых лиц на участие в испытаниях и сбора данных о побочных действиях препаратов.
Внедрение правил GСР позволяет:
-
улучшить методологию клинических испытаний и получить более надежные ре-зультаты исследований относительно эффективности и безопасности;
-
гарантировать защиту интересов участников испытаний;
-
ускорить разработку новых препаратов и, следовательно, доступ пациентов к но-вым лекарствам;
-
ускорить спонсорам (разработчикам/производителям) выход к новым рынкам;
-
клиническим учреждениям участвовать в многоцентровых международных клини-ческих испытаниях.
-
Соблюдение требований GСР содействует получению объективных результатов в качестве основы фармакотерапии, оказывает положительное влияние на использование лекарственных средств и на медицинскую практику в целом.
-
При применении правил GCP возрастает роль проверяющих (инспектирующих) инстанций, повышаются требования к клиницистам, участвующим в испытаниях, и к при-борному оснащению клиник.
27. Перечислите основные требования правил gmp к производству лекарственных средств.
Основные требования GMP:
I. Все производственные процессы должны быть четко регламентированы и должны периодически пересматриваться с учетом накопленного опыта. Следует контролировать стабильность производства лекарственных средств с заданным качеством в соответствии со спецификациями на них.
-
Следует проводить аттестацию (испытания) критических стадий процессов про-изводства, в том числе при внесении существенных изменений в технологический про-цесс.
-
Следует обеспечить все необходимые условия для выполнения требований настоящего стандарта, в т.ч., включая наличие:
- обученного и аттестованного персонала; - необходимых помещений и площадей;
- соответствующего оборудования и системы обслуживания;
- материалов, средств упаковки и маркировки, удовлетворяющих установленным требованиям;
- утвержденных инструкций и методик;
- требуемых условий хранения и транспортирования.
IV. Инструкции и методики должны быть конкретными, изложены ясно и однознач-но в письменной форме.
V. Персонал должен быть обучен правильному выполнению инструкций.
VI. В процессе производства следует составлять протоколы (заполняемые рукопис-ным способом и/или с применением технических средств), документально подтверждаю-щие фактическое проведение предусмотренных инструкциями технологических стадий и получение продукции требуемого качества в количестве, соответствующем установленным нормам. Все отклонения необходимо расследовать и протоколировать в полном объеме.
VII. Протоколы на серию продукции, в т.ч. на документацию по реализации продук-ции, должны давать возможность прослеживать изготовление каждой серии и храниться в полном объеме в доступной форме.
VIII. Порядок реализации (оптовой продажи) продукции должен сводить к миниму-му любой риск для ее качества.
IX. Следует организовать систему отзыва любой серии продукции из продажи или поставки.
X. Рекламации на качество продукции следует тщательно рассматривать, а причины ухудшения качества расследовать с принятием соответствующих мер по их предотвраще-нию.
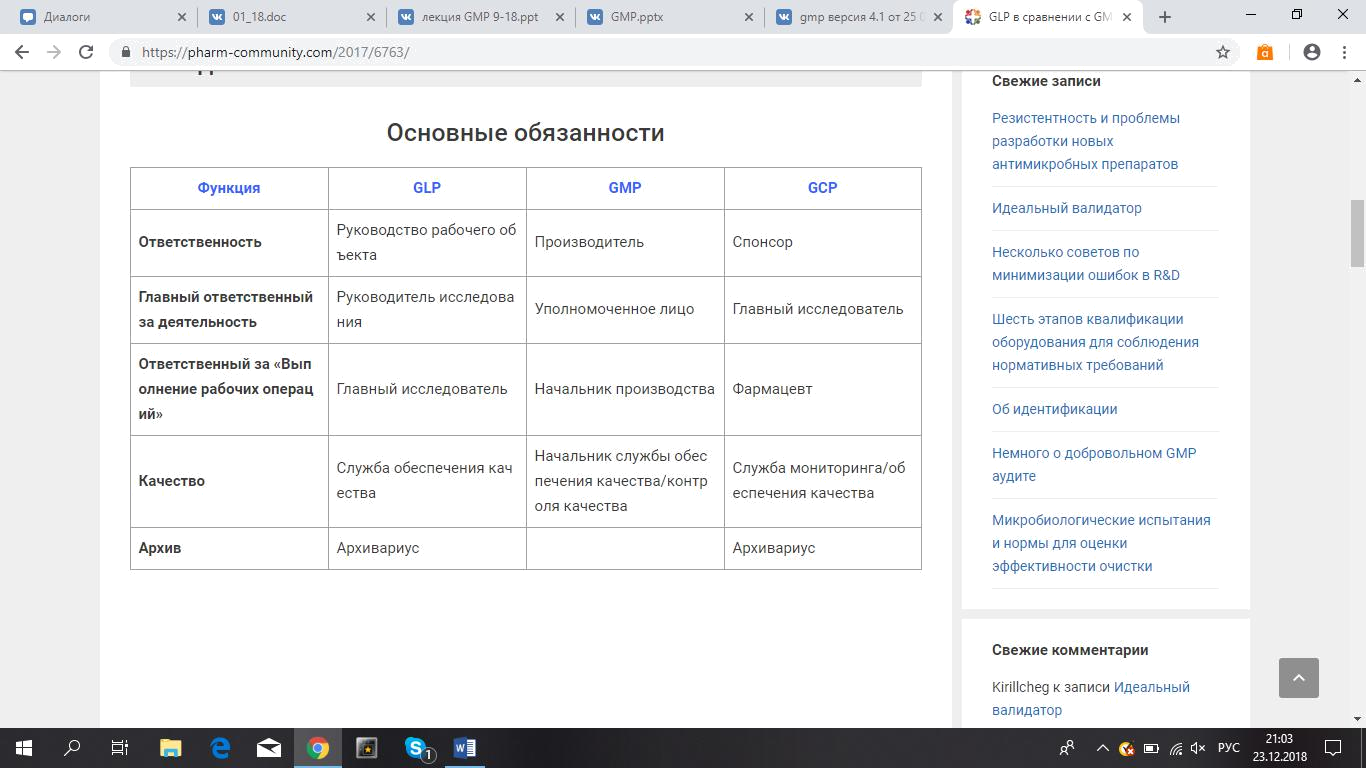
28. Приведите различия и общие принципы правил gmp, gcp и glp.
Надлежащая лабораторная практика (GLP)
-
Система качества, охватывающая организационный процесс и условия, при которых планируют, проводят, мониторят, ведут учет, архивируют и сообщают доклини-ческие исследования по безопасности для здоровья и окружающей среды.
-
GLP представляет собой нормативные требования, опубликованные в Своде федеральных постановлений США (CFR) (21 CFR, часть 58)
-
Принципы Организации экономического сотрудничества и развития (ОЭСР)
GLP
-
GLP не является руководством или рекомендацией, она носит законодатель-ный характер.
-
GLP не является «урезанной» GMP!
Надлежащая производственная практика
-
Надлежащая производственная практика является частью системы обеспе-чения качества, гарантирующей постоянство (стабильность) производства и контроля продукции по стандартам качества в соответствии с её предусмотренным применением и требованиями регистрационного удостоверения или спецификации продукта.
-
GMP охватывает как контроль производства, так и контроль качества
-
Правила, регулируют лекарственные препараты в ЕС; Том 4 – Руководства ЕС по GMP для медицинских и ветеринарных препаратов
Надлежащая клиническая практика
-
GCP является международным этическим и научным стандартом качества по планированию, проведению, учету и сообщению исследований с участием человека.
-
Соблюдение требований данного стандарта предоставляет общественные гарантии того, что права, безопасность и благополучие субъектов исследования защище-ны, согласуются с принципами, изложенными в Хельсинкской декларации, и то, что кли-нические данные являются достоверными.
-
Руководством по Надлежащей клинической практике является гармонизиро-ванное трехстороннее руководство Международной конференции по гармонизации (ICH)
-
Правила GLP не применяются к какой-либо стадии производства
-
Продукт, произведенный для токсикологических исследований, должен быть ис-следован на безопасность. Эти in vitro и in vivo исследования должны быть проведены в соответствии с требованиями GLP
-
Клинический материал должен быть исследован на безопасность и эффективность
-
участием людей. Эти исследования с участием людей должны проводиться в соответ-ствии с требованиями GCP
29. Требования к качеству лекарственных веществ, вспомогательных веществ и материалов.
Лекарственные средства — вещества или их комбинации, вступающие в контакт с орга-низмом человека или животного, проникающие в органы, ткани организма человека или
28
животного, применяемые для профилактики, диагностики (за исключением веществ или их комбинаций, не контактирующих с организмом человека или животного), лечения за-болевания, реабилитации, для сохранения, предотвращения или прерывания беременности
-
полученные из крови, плазмы крови, из органов, тканей организма человека или живот-ного, растений, минералов методами синтеза или с применением биологических техноло-гий. К лекарственным средствам относятся фармацевтические субстанции и лекарствен-ные препараты.
Вспомогательные вещества — вещества неорганического или органического происхожде-ния, используемые в процессе производства, изготовления лекарственных препаратов для придания им необходимых физико-химических свойств.

29

30. Требования gmp к процессу производства готовых лекарственных препаратов.
Принцип: Технологические операции должны осуществляться по четко установлен-ным процедурам; они должны отвечать настоящим Правилам для получения продукции требуемого качества и соответствовать лицензии на производство и регистрационному досье.
Общие требования
5.1. Производственный процесс должен осуществляться и контролироваться квали-фицированным персоналом.
5.2. Все действия, проводимые с материалами и продукцией, такие как приемка и ка-рантин, отбор проб, хранение, маркировка, выдача в производство, технологический про-цесс, упаковка и реализация, следует осуществлять согласно письменным процедурам или инструкциями оформлять документально.
5.3. Все поступающие материалы должны быть проверены, чтобы гарантировать, что поставка соответствует заказу. Тара должна быть очищена (при необходимости) и марки-рована с указанием требуемой информации.
5.4. Факты повреждения тары и упаковки и любые другие проблемы, которые могут неблагоприятно повлиять на качество материалов, должны быть расследованы, оформле-ны документально, а информация о них должна быть доложена в подразделения контроля качества.
5.5. Поступающие материалы и произведенная готовая продукция должны немед-ленно помещаться в карантин, действующий по принципу раздельного хранения или за счет организационных мер, и содержаться в нем до получения разрешения на использова-ние или реализацию.
5.6. Приемку закупаемых промежуточной и нерасфасованной продукции проводят в соответствии с правилами, действующими для исходных материалов.
5.7. Все материалы и продукцию следует хранить в соответствующих условиях, установленных производителем, в определенном порядке, обеспечивающем разделение по сериям и установленную очередность использования складских запасов.
31. Способы очистки и стерилизации воздуха. Механизмы осаждения частиц аэрозоля на волокнистых фильтрах. Классификация воз-
душных фильтров.
-
Высокотемпературная обработка воздуха
-
Ультрафиолетовое излучение
-
Химическая стерилизация
-
Фильтрующая стерилизация
Фильтрование запыленного потока через слой пористого материала - сложный про-цесс, включающий действие ситового эффекта, инерционного столкновения, броуновской диффузии, касания (зацепления), действия гравитационных и электрических сил.
При приближении частицы к волокну действует несколько механизмов, которые мо-гут привести к ее улавливанию:
-
касание;
-
инерционный захват;
-
диффузия;
-
ситовой эффект.
Осаждение частиц на поверхности пор фильтрующего элемента происходит в ре-зультате совокупного действия эффекта зацепления, а также диффузионного, инерционно-го и механизмов. Пыль при фильтровании в основном задерживается в результате столк-новения частиц с волокнами и нитями фильтровального материала и прилипания частиц к волокнам.
-
Касание. Частица переносится вдоль линии тока газа к нити или волокну (препят-ствию). Если частица движется мимо препятствия на расстоянии меньше своего радиуса, то она касается препятствия и захватывается.
-
Инерция. Частица находится на линии тока, следуя которой она прошла бы мимо препятствия, не касаясь его, но под действием инерции частица сходит с первоначальной линии тока. В результате она сталкивается с препятствием. Чем больше частица, тем больше ее инерция, лучше условия для захвата. При обычных скоростях течения в филь-трах этот механизм мало эффективен для частиц диаметром менее микрометра.
-
Диффузия. Частица настолько мала, что ее траектория становится хаотичной из-за броуновского движения. Захват может произойти, если случайное отклонение приводит частицу к волокну. Этот механизм становится наиболее важным, когда размер частиц меньше 0,1 мкм.
-
Ситовой эффект. Частица задерживается из-за того, что слишком велика, чтобы пройти через данную пору или канал.
Возможности осаждения за счет ситового эффекта, особенно при прохождении по-тока через чистую ткань, ограничены, т. к. в большинстве случаев размеры частиц значи-тельно меньше размеров пор.
Классификация воздушных фильтров по европейским стандартам EN 779, EN
1822 И гост р 51251-99:
Воздушные фильтры в соответствии со своими характеристиками разделены на 4 группы:
-
Фильтры грубой очистки – G
-
Фильтры тонкой очистки – F
-
HEPA (High Efficiency Particulate Air) – H
-
ULPA (Ultra Low Penetration Air) - U
Классификация воздушных фильтров общего назначения:
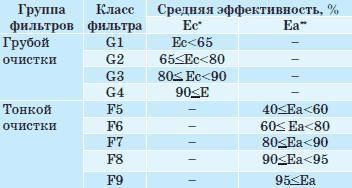
-
- Определеяется по синтетической пыли.
-
- Определеяется для частиц 0,4 мкм
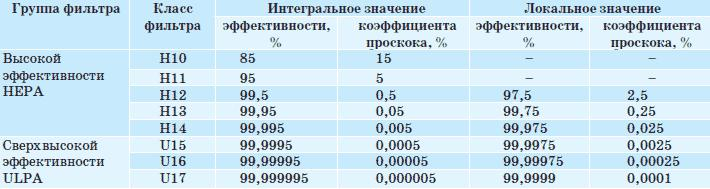
Классификация высоко- (НЕРА) и сверхвысокоэффективных (ULPA) воздушных фильтров: