Материал: GMP_prefinal
-
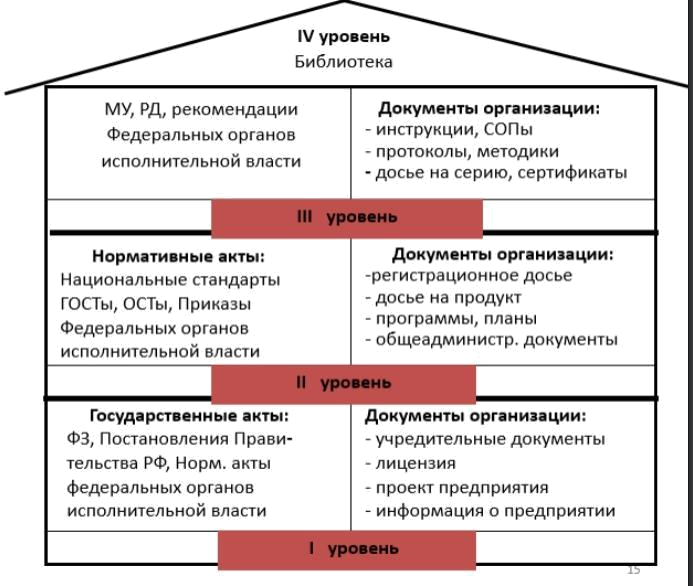
Нормативная документация, регламентирующая производство и качество лекарственных препаратов.
Производство:
-
ФЗ «О техническом регулировании»
-
ФЗ «Об обращении ЛС»
Также:
-
Технологические регламенты.
-
Государственная фармакопея (ГФ).
-
Фармакопейные статьи (ФС).
-
Временные фармакопейные статьи (ВФС).
-
Государственные стандарты (ГОСТ).
-
Отраслевые стандарты (ОСТ).
-
Технические условия (ТУ).
-
Руководящий нормативный документ (РД) – инструкции, методические указания и т. д.
-
Производственные и технологические инструкции.
12. Охарактеризуйте систему стандартов gxp.
GxP - Система надлежащих практик (Good … Practice), система специальных отраслевых стандартов, принятых в мировой фармацевтической отрасли, устанавливающих требования к оборудованию, персоналу, обеспечению и контролю качества, полупродуктам, продуктам, а также результатам деятельности компаний на всех этапах обращения лекарственных средств.
Русский Регистр является единственной в России организацией, имеющей государственную аккредитацию (Федеральная служба по аккредитации РФ) на право сертификации в соответствии со стандартами надлежащих практик
-
GLP - правила доклинических исследований
-
GCP - правила клинических исследований
-
GМP - правила надлежащего производства ЛС
-
GDP – правила оптовой торговли лекарственными средствами
-
GPP – правила розничной торговли лекарственными средствами
-
GSP - правила надлежащего хранения ЛС
-
GRP - правила надлежащего найма на работу с ЛС
13. Опишите нормативно–правовую базу лицензирования производства лекарственных средств в рф.
Лицензирование производства лекарственных средств фармацевтической деятельности осуществляется в соответствии с законодательством Российской Федерации.
Постановление Правительства РФ от 06.07.2012 N 686 (ред. от 20.06.2018) "Об утверждении Положения о лицензировании производства лекарственных средств»
Нормативно-правовая база, регламентирующая предоставление лицензии на фармацевтическую деятельность:
- Федеральный закон № 99 от 04.05.2011 г. «О лицензировании отдельных видов деятельности»;
-
ФЗ от 22.08.2004 г. № 122-ФЗ «О лекарственных средствах»;
- ФЗ от 08.01.1998 г. № 3-ФЗ «О наркотических средствах и психотропных веществах»;
- Постановление Правительства РФ от 06.07.2006 г. № 416 «Об утверждении Положения о лицензировании фармацевтической деятельности» (ред. от 19.07.2007 г.);
- Постановление Правительства РФ от 04.11.2006 г. № 648 «Об утверждении Положения о лицензировании деятельности, связанной с оборотом наркотических средств и психотропных веществ»;
- Постановление Правительства РФ от 06.07.2006 г. № 415 «Об утверждении Положения о лицензировании производства лекарственных средств»;
- Постановление Правительства РФ от 06.07.2005 г. № 438 «О порядке ввоза и вывоза лекарственных средств, предназначенных для медицинского применения»;
-
Приказ Минздрава России от 04.03.2003 г. № 80 «Об утверждении отраслевого стандарта "Правила отпуска (реализации) лекарственных средств в аптечных организациях. Основные положения"»;
-
Приказ МЗСР РФ от 12.02.2007 г. № 110 «О порядке назначения и выписывания лекарственных средств, изделий медицинского назначения и специализированных продуктов лечебного питания».
14. Перечислите и обоснуйте основные лицензионные требования и условия при осуществлении деятельности по производству лекарственных средств.
Федеральный закон от 12.04.2010 N 61-ФЗ«Об обращении лекарственных средств»
Глава 8. Производство и маркировка лекарственных средств
Статья 45. Производство лекарственных средств
1. Производство лекарственных средств должно соответствовать правилам организации производства и контроля качества лекарственных средств, утвержденным Правительством Российской Федерации.
2. Производство лекарственных средств на территории Российской Федерации осуществляется производителями лекарственных средств, имеющими лицензию на производство лекарственных средств.
3. Производство лекарственных средств осуществляется с соблюдением требований промышленного регламента, который утверждается руководителем производителя лекарственных средств и включает в себя перечень используемых фармацевтических субстанций и вспомогательных веществ с указанием количества каждого из них, данные об используемом оборудовании, описание технологического процесса и методов контроля на всех этапах производства лекарственных средств.
4. При производстве лекарственных средств используются фармацевтические субстанции, включенные в государственный реестр лекарственных средств.
-
Лицензирование деятельности по производству лекарственных средств для медицинского применения осуществляет Министерство промышленности и торговли Российской Федерации, для ветеринарного применения - Федеральная служба по ветеринарному и фитосанитарному надзору
-
Требования:
-
наличие у соискателя лицензии помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, принадлежащих ему на праве собственности или на ином законном основании, необходимых для выполнения за-являемых работ, соответствующих установленным требованиям;
-
соответствие производства лекарственных средств правилам организации производства и контроля качества лекарственных средств в соответствии со статьей 45 Федерального закона "Об обращении лекарственных средств";
-
наличие в соответствии со статьей 45 Федерального закона "Об обращении лекар-ственных средств" промышленных регламентов, утвержденных руководителем произво-дителя лекарственных средств (соискателя лицензии) и включающих в себя перечень ис-пользуемых фармацевтических субстанций и вспомогательных веществ с указанием коли-чества каждого из них, данные об используемом оборудовании и описание технологиче-ского процесса и методов контроля на всех этапах производства лекарственных средств;
-
наличие в соответствии со статьей 45 Федерального закона "Об обращении лекар-ственных средств" уполномоченного лица производителя лекарственных средств, которое при вводе лекарственных средств в гражданский оборот осуществляет подтверждение со-ответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с правилами производства и контроля качества лекарственных средств, а также которое:
-
имеет высшее фармацевтическое, химическое, медицинское или био-логическое образование либо при производстве лекарственных средств для ветеринарного применения - ветеринарное образование, стаж работы не менее чем 5 лет в области произ-водства и контроля качества лекарственных средств;
-
при производстве лекарственных средств для медицинского применения аттестовано в порядке, установленном Министерством здравоохранения Российской Федерации, при производстве лекарственных средств для ветеринарного применения - в порядке, установленном Министерством сельского хозяйства Российской Федерации;
-
наличие работников, заключивших трудовые договоры, имеющих соответственно высшее или среднее профессиональное фармацевтическое, химическое, химико-технологическое, биологическое, биотехнологическое, медицинское или ветеринарное об-разование, ответственных за производство и маркировку лекарственных средств.
-
Соблюдение лицензиатом требований о запрещении производства лекарственных средств, не включенных в государственный реестр лекарственных средств, за исключением лекарственных средств, производимых для проведения клинических исследований и экспорта, а также о запрещении производства, фальсифицированных лекарственных средств;
-
Соблюдение лицензиатом требований о запрещении продажи недоброкачественных лекарственных средств, фальсифицированных лекарственных средств и контрафактных лекарственных средств;
-
Соблюдение лицензиатом порядка уничтожения недоброкачественных лекарственных средств, фальсифицированных лекарственных средств и контрафактных лекарственных средств (утвержден постановлением Правительства Российской Федерации от 3 сентября 2010 г. № 674);
-
Соблюдение лицензиатом требования о государственной регистрации установленных производителями лекарственных препаратов предельных отпускных цен на лекарственные препараты, включенные в перечень жизненно необходимых и важнейших лекарственных препаратов.
-
Осуществление деятельности по производству лекарственных средств с грубым нарушением лицензионных требований влечет за собой ответственность, установленную законодательством Российской Федерации.
15. Опишите порядок государственной регистрации лекарственных средств в рф. Охарактеризуйте препараты, подлежащие и не подлежащие государственной регистрации в рф.
К заявлению о предоставлении (переоформлении) лицензии прилагаются:
-
копии учредительных документов юридического лица, засвидетельствованные в нотариальном порядке;
-
оригинал лицензии (при переоформлении лицензии);
-
документ, подтверждающий уплату государственной пошлины за предоставление лицензии;
-
копии документов, перечень которых определяется положением о лицензировании конкретного вида деятельности и которые свидетельствуют о наличии у соискателя лицензии возможности выполнения лицензионных требований и условий, а именно: копии документов, свидетельствующих о наличии у соискателя лицензии (лицензиата) необходимых для осуществления лицензируемой деятельности зданий, помещений и оборудования, принадлежащих ему на праве собственности или на ином законном основании, либо документов, подтверждающих наличие законных оснований для использования помещений и оборудования или только оборудования, принадлежащих организациям-производителям лекарственных средств (с представлением оригиналов в случае, если верность копий не засвидетельствована в нотариальном порядке); перечень работ по производству лекарственных форм и (или) видов фармацевтических субстанций, которые заявитель намерен производить (наименование лекарственной формы приводится в соответствии с наименованиями форм, перечисленными в Государственном реестре лекарственных средств). Возможность производства каждой лекарственной формы должна быть подтверждена наличием соответствующих помещений и оборудования.
-
копии документов, подтверждающих соответствующую лицензионным требованиям и условиям квалификацию уполномоченных лиц, а также специалистов, ответственных за производство и маркировку лекарственных средств. Рекомендуемая форма сведений о квалификации специалистов, ответственных за производство, качество и маркировку лекарственных средств, приведена на сайте. До вступления в действие нормативных правовых актов об уполномоченном лице предоставляются сведения о лице, ответственном за качество лекарственных средств.
-
По усмотрению заявителя к заявлению могут быть приложены прочие документы, подтверждающие возможность выполнения заявителем лицензионных требований и условий, но не входящие в перечень (например, информация о предприятии (мастер-файл), свидетельствующая о наличии у лицензиата/соискателя лицензии возможности выполнения лицензионных требований и условий, копии выданных сертификатов GMP и др.)
-
Рекомендуемая форма заявления
www.minpromtorg.gov.ru/special/medicines/0/zayavlenie_MP.doc
-
Регистрация лекарственных средств в России является одним из компонентов системы защиты здоровья и жизни населения от небезопасных, неэффективных и некачественных лекарственных средств (ЛС).
-
Регистрация лекарственных средств используется государствами во всех странах мира и осуществляется государственными органами исполнительной власти в области здравоохранения.
-
В соответствии с Главой 6. «Осуществление государственной регистрации лекарственных препаратов» Федерального закона “Об обращении лекарственных средств” от 12.04.2010г. № 61-ФЗ, в Российской Федерации для использования в медицинской практике лекарственных средств (как отечественных, так и зарубежных) необходима их государственная регистрация.
Государственной регистрации в РФ подлежат:
-
новые лекарственные средства;
-
новые комбинации зарегистрированных ранее лекарственных средств;
-
лекарственные средства, зарегистрированные ранее, но произведенные в других лекарственных формах, с новой дозировкой или другим составом вспомогательных ве-ществ;
-
воспроизведенные лекарственные средства.
Государственной регистрации не подлежат:
-
лекарственные препараты, изготовленные аптечными организациями, ветери-нарными аптечными организациями, индивидуальными предпринимателями, которые имеют лицензию на фармацевтическую деятельность, по рецептам на лекарственные препараты и требованиям медицинских организаций, ветеринарных организаций;
-
лекарственное растительное сырье;
-
лекарственные препараты, приобретенные физическими лицами за пределами тер-ритории Российской Федерации и предназначенные для личного использования;
-
лекарственные препараты, предназначенные для экспорта;
-
радиофармацевтические лекарственные препараты, изготовленные непосредствен-но в медицинских организациях в порядке, установленном уполномоченным федеральным органом исполнительной власти.
Государственная регистрация лекарственных препаратов осуществляется соответ-ствующим уполномоченным федеральным органом исполнительной власти в срок, не превышающий 210 рабочих дней со дня принятия заявления о государственной регистрации лекарственного препарата. В указанный срок включается время, необходимое для повтор-ного проведения экспертизы лекарственных средств и (или) этической экспертизы в соот-ветствии со статьей 25 настоящего Федерального закона.
Срок государственной регистрации лекарственного препарата исчисляется со дня принятия соответствующим уполномоченным федеральным органом исполнительной вла-сти заявления о государственной регистрации лекарственного препарата с приложением необходимых документов по день выдачи регистрационного удостоверения лекарственного препарата.
Время проведения клинического исследования лекарственного препарата не учитывается при исчислении срока его государственной регистрации.
Государственная регистрация лекарственных препаратов для медицинского приме-нения осуществляется по результатам экспертизы лекарственных средств и этической экспертизы возможности проведения клинического исследования лекарственного препа-рата для медицинского применения (далее - этическая экспертиза).
Государственная регистрация лекарственных препаратов для ветеринарного приме-нения осуществляется по результатам экспертизы лекарственных средств для ветеринарного применения.
ПОРЯДОК РЕГИСТРАЦИИ:
1. Составление регистрационного досье, включая документы необходимые для начала клинического исследования, и сдача досье в Минздравсоцразвития России.
-
Данные об активных фармацевтических субстанциях, используемых для производства лекарственного препарата
III. Описание фармацевтических свойств готового лекарственного препарата IV. Данные о производстве лекарственного препарата
V. Данные по контролю качества лекарственного препарата
VI. Сведения о результатах доклинических фармакологических и токсикологических исследований лекарственного препарата
VII. Сведения о результатах клинических исследований лекарственного препарата
-
Получение разрешения на проведение клинического исследования и его проведение в РФ.
-
Экспертиза качества лекарственного препарата и экспертиза отношения ожидае-мой пользы к возможному риску применения лекарственного препарата, осуществляемые после проведения его клинического исследования: