Материал: Шмид Р. Наглядная биотехнология и генетическая инженерия
развития |
Поиск биологически активных веществ |
||
ВВЕДЕНИЕ. Поиск новых лекарств или химикатов для |
в 96или 384-гнездных микротитровальных планше- |
||
|
|||
Тенденции |
сельскохозяйственных нужд традиционно проводился |
тах. Использование реакционных камер с объемом |
|
ствуют в организме на определенный белок (или бел- |
НЫХ ВЕЩЕСТВ. Несмотря на то что пространственные |
||
|
методом проб и ошибок. Естественно-научные знания |
~10–9 л на основе травленых кремниевых плат в со- |
|
|
и новые технологии позволяют сделать этот поиск |
четании с конфокальной лазерной спектроскопией |
|
|
более рациональным. В основе такого подхода лежит |
позволяют провести более 10 000 измерений в день. |
|
|
идея о том, что биологически активные вещества дей- |
РАЦИОНАЛЬНЫЙ ДИЗАЙН БИОЛОГИЧЕСКИ АКТИВ- |
|
|
ки), называемые мишенями. В роли мишеней могут |
структуры большинства белков-мишеней неизвест- |
|
|
выступать ферменты, рецепторы, белки ионных кана- |
ны, делаются попытки поиска сайтов связывания ми- |
|
|
лов, в случае растений – белки, участвующие в фото- |
шеней с биологически активными веществами. Для |
|
|
синтезе. Анализ генома и применение методов |
этого исследуют взаимодействие мишеней с различ- |
|
|
протеомики позволяют значительно эффективнее |
ными синтетическими аналогами природных субстра- |
|
|
проводить поиск мишеней. Методами генетической |
тов (QSAR – quantative structure – activity relations- |
|
|
инженерии можно выделить эти белки, а затем изу- |
hip). Полученные данные помогают изучать структуру |
|
|
чить их взаимодействие с природными и синтетиче- |
биологически активного вещества и усовершенство- |
|
|
скими веществами. На основе полученных данных о |
вать его в соответствии с поставленными целями |
|
|
структуре действующего вещества в дальнейшем воз- |
(drug design). |
|
|
можно создание модифицированных соединений, об- |
ПЕРСПЕКТИВЫ. На сегодняшний день из 50 000 ис- |
|
|
ладающих определенной биологической активностью |
следованных соединений лишь одно доходит до ста- |
|
|
и некоторыми дополнительными свойствами. Скриниг |
дии лекарственного препарата. В год в мире появля- |
|
|
с применением методов химической комбинаторики и |
ется около 30 новых фармацевтических продуктов. |
|
|
современных биохимических данных позволяет полу- |
Специалисты в области фармакологии надеются, что |
|
|
чать новые лекарственные вещества. |
с применением направленного скрининга это число |
|
|
ИДЕНТИФИКАЦИЯ И ПОЛУЧЕНИЕ МИШЕНЕЙ. Несмот- |
значительно возрастет. Анализ связанных с патоло- |
|
|
ря на значительный прогресс биотехнологии в пос- |
гией полиморфизмов должен выявить возможные |
|
|
ледние десятилетия, идентификация мишеней, в осо- |
индивидуальные отклонения в структуре гена-мише- |
|
|
бенности в случае полигенных заболеваний, остается |
ни, вызывающие те или иные проявления заболева- |
|
|
весьма сложной задачей. Исследование генофонда |
ния. Эта информация поможет устанавливать диаг- |
|
|
замкнутых популяций, например в Исландии или Тас- |
ноз на ранних стадиях заболевания и подбирать |
|
|
мании, и изучение геномов больных в этих популя- |
лекарственные средства, наиболее подходящие для |
|
|
циях способствовали определенному прогрессу в |
каждого конкретного пациента (фармакогеномика). |
|
|
этой области. Если удалось установить связь между |
|
|
|
дефектом фермента, ионного канала или рецептора и |
|
|
|
проявлениями заболевания, на следующем этапе по- |
|
|
|
лучают лабораторное животное, в котором «подозре- |
|
|
|
ваемый» ген выключен, например, с помощью анти- |
|
|
|
смысловой РНК. К примеру, если G-связанный |
|
|
|
рецептор распознается как мишень, он может быть |
|
|
|
локализован в мембране фибробластов мыши. В ка- |
|
|
|
честве цитоплазматического репортера используют |
|
|
|
экспрессию люциферазу светляка. Как только лиганд |
|
|
|
связывается с рецептором, происходит передача сиг- |
|
|
|
нала путем повышения концентрации цАМФ в цито- |
|
|
|
плазме, что приводит к развитию доступного для ко- |
|
|
|
личественной оценки люминесцентного сигнала при |
|
|
|
наличии субстрата люциферазы – люциферина. |
|
|
|
МАСШТАБНЫЙ СКРИНИНГ. Наиболее распространен- |
|
|
|
ный современный метод масштабного скрининга за- |
|
|
|
ключается в исследовании действия веществ из об- |
|
|
|
ширных библиотек химических соединений (более |
|
|
|
100 000 соединений) на мишень. Получение библи- |
|
|
|
отек методом химической комбинаторики позволяет |
|
|
270 |
практически безгранично увеличить количество ве- |
|
|
ществ в библиотеке. Такого рода измерения проводят |
|
||
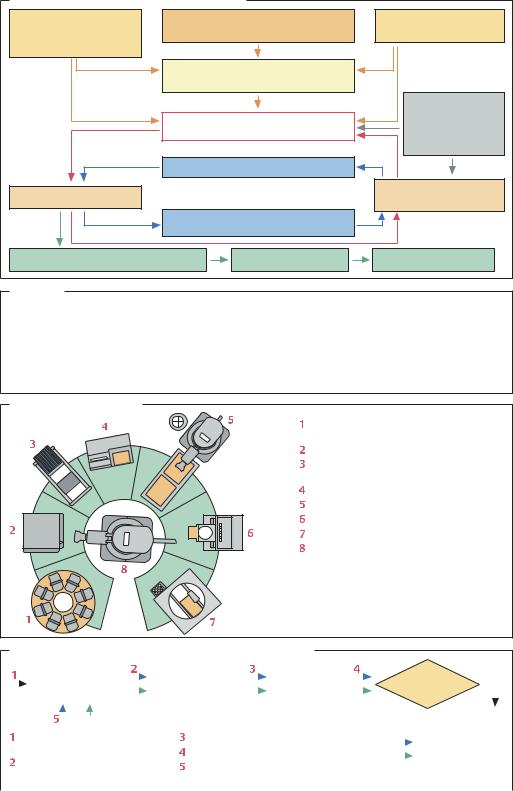
Скрининг биологически активных веществ |
|
|||
Природные вещества, |
Биологические мишени, например, |
Литературные данные, |
||
пептиды, синтетические |
||||
из анализа генома или протеома |
вещества-аналоги |
|||
вещества, комбинаторная |
||||
|
|
|
||
химия |
|
|
|
|
|
Экспериментальная модель, |
|
||
|
масштабный скрининг |
|
||
|
|
Основная структура |
Фармацевтические |
|
|
|
требования, |
||
|
|
активного вещества |
||
|
|
побочные явления |
||
|
|
|
||
|
Концепция эксперимента, синтеза |
|
||
Биологическая проверка |
|
|
Определение структуры, |
|
|
|
компьютерный дизайн |
||
|
|
|
||
|
Установление связи между структурой |
|
||
|
и действием вещества, QSAR |
|
||
Связь с интересующими свойствами |
Выбранное вещество |
Лекарственный препарат |
||
Мишени |
|
|
|
|
|
Количество |
Пример |
Применение |
|
в организме человека |
|
|
Ферменты |
8 000 |
Ацетилхолинэстераза |
Болезнь Альцгеймера |
Рецепторы |
15 000 |
Рецептор серотонина |
Шизофрения |
Ионные каналы |
3 000 |
Са-канал |
Болезни старческого возраста |
|
|
|
|
Масштабный скрининг |
Клетки-мишени на микротитровальном |
планшете |
СО2-инкубатор* |
Добавление веществ–кандидатов |
и реагентов |
Анализатор |
Автоматизированное нанесение проб |
Отмывка планшета |
Центрифугирование планшета |
Автоматизированное управление |
процессом |
* Необходим только при использовании |
в качестве мишеней культур клеток |
человека или животных |
Рациональный дизайн биологически активных веществ |
|
|
|
Трехмерная |
|
Предложенный |
|
|
|
Синтетические |
|
|
|
Биологически |
|
|
|
|||||||||
|
|
|
структура или |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
структурная модель |
|
|
|
лиганд |
|
|
|
соединения |
|
|
|
|
активен? |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Отходы |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
Определение структуры |
Химический синтез лигандов |
|
|
|
Цикл 1 |
|
|
|
|
||||||||||||||
|
|
белка или создание модели |
Биологический тест |
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
Цикл 2 |
и т. д.: |
|||||||||||||||||||
|
|
Дизайн лигандов |
Определение структуры или |
|
|
|
|||||||||||||||||||
|
|
|
|
|
оптимизация |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
создание модели мишени и лиганда |
|
|
|
|
|
|
|
|
|
|
271 |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
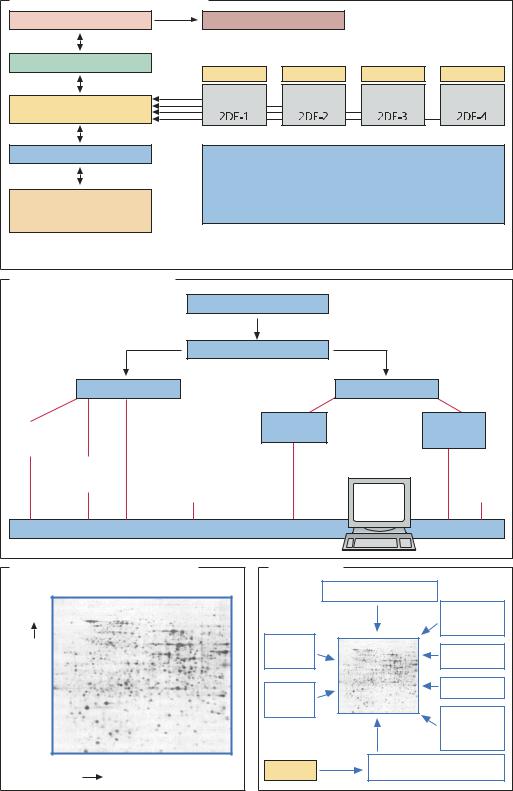
Один геном – различные протеомы |
|
|
|
|
Геномная библиотека |
Геном |
|
|
|
Библиотека мРНК |
Протеом 1 |
Протеом 2 |
Протеом 3 |
Протеом 4 |
|
||||
Протеомная библиотека |
|
|
|
|
Белковая библиотека |
|
Анализ белка |
|
|
|
• Определение N-концевых |
• Масс-спектрометрия |
||
|
аминокислот |
|
• Посттрансляционные |
|
|
• Аминокислотный состав |
модификации |
|
|
Литературные, |
• Расщепление |
|
• Иммуноанализ (вестерн-блот, |
|
биологические, |
протеиназами |
|
белковые микрочипы) |
|
медицинские данные |
|
|
|
|
Высший организм с 30 000 генов содержит вплоть до 1 000 000 белков, |
|
|
||
уровень экспрессии которых зависит от типа клеток (10 000 белков на каждый тип клеток) |
|
|||
Методы протеомного анализа |
|
|
|
|
|
Приготовление пробы |
|
|
Электроблоттинг |
Разделение белков |
Обработка |
|
|
|
||
|
|
трипсином в геле |
|
|
|
|
|
Интактные белки |
|
Пептидные фрагменты |
|
Анализ нуклео- |
|
Без |
Разделение |
|
разделения |
методами |
|
тидной последо- |
|
||
|
|
ЖХ* и КЭ** |
|
вательности |
|
|
|
|
|
|
|
Анализ амино- |
|
|
Анализ нук- |
кислотной пос- |
Методы масс-спектрометрии: |
|
леотидной |
ледовательности |
|
последова- |
|
|
MALDI-TOF, ESI-TOF и др. |
|
тельности |
|
Банки данных ДНК и белков |
|
|
* ЖХ – жидкостная хроматография. ** КЭ – капиллярный электрофорез. |
|
||
Двумерный электрофорез белков |
Применение |
|
|
пекарских дрожжей |
|
|
Обмен веществ |
100 кДа |
|
|
|
|
|
Взаимо- |
|
|
|
|
|
масса |
|
|
действия |
|
Лекарства |
Температура |
|
|
|
||
Молекулярная |
|
|
|
|
Химикаты |
Стресс |
|
|
|
||
|
|
|
|
|
|
|
Взаимо- |
|
|
|
действие |
|
|
|
с другими |
6 кДа |
|
|
клеткам |
|
|
Экспрессия, специфическая |
|
|
|
Геном |
|
Изоэлектрическая точка |
для данного типа клеток |
||
|
273 |
||
|
|
|
|
