Материал: Огурцов А. Н. Методы бииоинформационного анализа
Методы слов, или k-кортежей, которые реализованы в алгоритмах таких программ, как FASTA и BLAST, являются достаточно быстрыми и позволяют просматривать полную базу данных при поиске последовательности-кандидата, которая даёт наилучшее выравнивание с последовательностью запроса.
Алгоритмы программ FASTA и BLAST являются эвристическими, то есть основанными на эмпирических методах машинного программирования, в которых решение находится по установленным опытным путём правилам и используется обратная связь для уточнения результата.
Основная операция при поиске в базе данных – выравнивание последовательности запроса с каждой последовательностью-кандидатом базы данных; и если эвристические методы позволяют выполнить эту операцию значительно быстрее, то лучше применять именно их, а не алгоритмические методы динамического программирования.
Программа FASTA обеспечивает как высокий уровень чувствительности, так и большую скорость поиска подобия. Чувствительность достигается за счёт реализованных в программе FASTA алгоритмов оптимизированного локального выравнивания и анализа матрицы замен.
Сначала FASTA подготавливает список слов-кортежей, выбранных из пары сравниваемых последовательностей. Слово представляет собой строку из 3-6 нуклеотидов или 1-2 аминокислот. При этом слова-кортежи не должны перекрываться. Затем программа сопоставляет слова-кортежи и ведёт подсчет совпадений.
Подобно построению диаграммы и вычислению счёта точечной матрицы, FASTA отбирает варианты с максимальным числом слов на диагонали, находит совпадение с возможно высоким счётом и помечает результат (совпадение слов) как элемент № 1. Если максимальный счёт оказывается достаточно большим, программа переходит ко второму уровню.
На втором уровне для каждого лучшего совпадения слов производится поиск соседних приблизительных совпадений, и если значение счёта удовлетворительно, то программа объединяет короткие
70
сегменты элемента № 1, строит из них более длинную диагональ точечной матрицы и вычисляет счёт после включения пропуска и оценки штрафа.
Наилучший счёт из счетов второго уровня называют начальным числом. FASTA сохраняет счета начальных чисел, вычисленные для всех сравнений последовательности запроса с предметной последовательностью. После того как все последовательности базы данных проверены, те последовательности, которые дают максимальные счета начальных чисел, используются для построения (с помощью алгоритма СмитаУотермена) локального выравнивания с возможно большим оптимальным счётом.
Файл данных в формате FASTA включает в себя строку-титр (заголовок) и строки данных о последовательности. Описание последовательности следует за строкой-титром, в начале которой стоит знак «>». Первое слово в этой строке – название последовательности, далее идёт описание последовательности. Остальные строки содержат саму последовательность. При чтении файла данных программа FASTA игнорирует пустые строки, а также все знаки пробелов или пропусков в последовательности. Файл, объединяющий в себе множество последовательностей, построен по тому же принципу – строки, содержащие описание последовательностей, идут друг за другом.
Формат FASTA принят во многих программах множественного выравнивания последовательностей.
3.5. ЗНАЧИМОСТЬ ВЫРАВНИВАНИЙ
Для оценки биологической значимости выравнивания данной последовательности с другими последовательностями из баз данных принято сравнивать полученные результаты с результатами выравнивания данной последовательности с последовательностями, полученными с помощью статистически случайных перестановок элементов в последовательностях из баз данных.
Очевидно, что если рандомизированные последовательности дают такой же результат, как и исходные, то, скорее всего, выравнивание не имеет биологического смысла.
71

Для оценки значимости выравниваний обычно используют такие статистические параметры, как Z-score, P-value и E-value.
Z-score показывает, насколько необычно обнаруженное нами совпадение, т. е. в терминах статистики – это расстояние (измеряемое как среднеквадратическое отклонение) данного уровня от среднего значения по набору данных. Если вес исходного выравнивания данной последовательности с другой последовательностью равен S, то
Z = S σ−μ,
где μ – среднее значение выравниваний данной последовательности с рандомизированными вариантами второй последовательности; σ – стандартное отклонение.
При Z = 0 для двух белковых последовательностей – эти белки похожи друг на друга, не сильней, чем (в среднем) на белки из некоторой контрольной группы, по которой и производится сравнение, что, впрочем, вполне может произойти случайно. Чем больше Z-score, тем больше вероятность того, что наблюдаемое выравнивание появилось неслучайно. Опыт показывает, что Z-score > 5 уже говорит о значимости исходного выравнивания.
P-value. Многие программы выдают величины Р (P-value) – вероятности того, что выравнивание не лучше, чем случайное. Связь Z-score и Р зависит от распределения весов контрольных выравниваний, которое не соответствует нормальному распределению.
Ориентировочно значения P-value можно интерпретировать следующим образом:
Р ≤10–100
10–100 < Р < 10–50
10–50 < Р < 10–10
точное совпадение;
последовательности почти идентичны, например, аллели или полиморфизмы;
близкородственные последовательности; гомология очевидна;
72
10–10 < Р < 10–1 |
обычно дальнеродственные последовательности; |
Р > 10–1 |
по-видимому, соответствие незначимо. |
E-value. Программы поиска по базам данных, в том числе и PSIBLAST, указывают E-value.
E-value выравнивания – это ожидаемое количество последовательностей, которые бы имели Z-score такой же (или лучше), как если бы мы в качестве запроса дали программе случайную последовательность.
Ориентировочно значения E-value можно интерпретировать следующим образом:
Е< 0,02 вероятно, последовательности являются гомологами; 0,02 < E < 1 гомология не очевидна;
Е> 1 следует ожидать, что это случайное совпадение.
Следует отметить, что статистические оценки полезны и необходимы, но они не могут заменить здравый смысл и тщательный и аккуратный анализ биологичности результатов.
Существует множество эмпирических правил интерпретации процента идентичных аминокислотных остатков в оптимальном выравнивании белковых последовательностей.
Если два белка содержат более 45% идентичных остатков в их оптимальном выравнивании, то есть все основания предполагать, что эти белки имеют подобные структуры и, скорее всего, общую или, по крайней мере, сходную функцию.
Если они содержат более 25% идентичных остатков, они, вероятно, имеют подобный фолдинг.
С другой стороны, низкая степень сходства последовательностей не может исключить возможность гомологии.
Р.Ф. Дулитл определил область 18-25% сходства последователь-
ностей как область двусмысленности (или, область неоднозначности),
для которой предположение о гомологии можно высказывать только в
73
качестве гипотезы. Парные выравнивания, которые находятся ниже этой области, малоинформативны.
При этом отсутствие значимого сходства последовательностей совсем не означает отсутствие сходства структур.
Хотя область неоднозначности и ненадежна для выводов, но для решения вопроса об истинном родстве важна также "текстура" (профили) выравнивания – изолированы ли эти сходные остатки и распределены по всей последовательности или же они образуют "айсберги" – локальные участки высокого сходства (ещё один термин Дулитла), которые могут соответствовать общему активному центру. Также полезно использовать дополнительную информацию об общих лигандах или функциях. В случае, если пространственные структуры известны, то мы можем проверить их сходство непосредственно.
Эмпирические правила являются скорее рекомендациями, чем закономерностями. Приведём несколько характерных примеров.
Миоглобин кашалота и леггемоглобин люпина имеют 15% иден-
тичных остатков в оптимальном выравнивании. Это даже ниже определенной Дулитлом области неоднозначности. Однако известно, что обе молекулы имеют сходные трехмерные структуры, содержат гемовые простетические группы и связывают кислород. Они действительно являются удаленными гомологами.
Последовательности N- и С-концевых частей в одном и том же белке роданез имеют 11% идентичных остатков в оптимальном выравнивании. Если бы они возникли в разных белках, нельзя было бы судить об их родстве, исходя лишь из последовательностей. Однако такая ситуация в одном белке дает основание полагать, что они произошли путём дупликации и дивергенции генов. Очевидное сходство их структур подтверждает их родство.
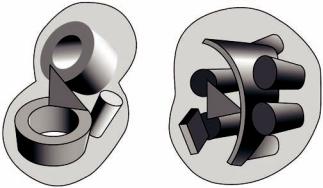
Две протеазы химотрипсин и субтилизин имеют последовательности схожие на 12%. Эти сериновые протеазы выполняют сходную функцию и их активный центр образован тремя характерными для них остатками. Тем не менее, они имеют разную пространственную укладку и не родственны (рисунок 20).
74
а |
б |
Рисунок 20 – Схема строения сериновых протеаз: а – типа трипсина; б – типа субтилизина. Схематически изображены α-спирали, β-листы и β-цилиндры. Район активного центра показан треугольником
Схожесть их каталитических функций – это пример конвергентной эволюции. Поэтому не стоит предполагать родственную связь между белками с непохожими последовательностями, основываясь только на схожести их функций.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1.Каковы недостатки метода динамического программирования?
2.За счёт чего сокращается время расчётов в методе динамического программирования?
3.Чем отличаются методы Нидлмена-Вунша и Смита-Уотермана?
4.Каков алгоритм заполнения ячеек F(i, j) матрицы динамического программирования?
5.Какие существуют два отличия алгоритма локального выравнивания от алгоритма глобального выравнивания?
6.Что такое k-кортеж?
7.Какие три статистических параметра используют для оценки значимости выравнивания?
75

4. МНОЖЕСТВЕННОЕ ВЫРАВНИВАНИЕ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Множественным называют выравнивание двух и более последовательностей. Групповой анализ последовательностей, входящих в семейства генов, предполагает установление связей между более чем двумя членами группы, что позволяет выявить скрытые консервативные характеристики семейства.
Цель множественного выравнивания последовательностей состоит в том, чтобы произвести краткую, но исчерпывающую характеристику данных о структуре последовательностей, на основании которой можно будет принять решение о принадлежности этих последовательностей к рассматриваемому семейству генов. По сравнению с попарным, множественное выравнивание даёт больше информации об эволюционной консервативности. Для того чтобы множественное выравнивание было максимально информативным, оно должно содержать равномерную выборку близко и отдалённо связанных последовательностей.
Для построения оптимального множественного выравнивания последовательностей в соответствующие столбцы сводят как можно больше подобных знаков. Множественное выравнивание группы последовательностей может обеспечить информацию о наиболее подобных областях, присущих этой группе. В белках такие области могут быть представлены консервативными доменами – функционально активными или структурными.
Если известна структура одного или нескольких членов выравнивания, то иногда возможно предсказать, какие аминокислоты образуют подобные пространственные структуры в других белках-членах выравнивания или какие гены занимают те же участки в последовательностях других нуклеиновых кислот-членов выравнивания. Множественное выравнивание последовательностей применяют также для
(1)предсказания зондов, специфичных к другим членам группы, или для
(2)открытия семейства подобных последовательностей – из одного или разных организмов.
76
4.1. ВИЗУАЛИЗАЦИЯ РЕЗУЛЬТАТОВ ВЫРАВНИВАНИЯ
Для облегчения анализа результатов множественного выравнивания белков аминокислотные остатки разных типов окрашивают на экране компьютера разными цветами. Один из возможных способов окраски представлен в таблице 10.
Таблица 10 – Один из возможных способов окраски аминокислотных остатков при визуализации множественного выравнивания белковых последовательностей
Цвет |
Тип остатка |
Аминокислоты |
|
|
|
|
|
Желтый |
Маленькие неполяр- |
Gly, Ala, Ser, Thr |
|
|
ные остатки |
|
|
Зеленый |
Гидрофобные |
Cys, Val, He, Leu, Pro, Phe, |
|
Tyr, Met, Trp |
|||
|
|
||
Фиолетовый |
Полярные |
Asn, Gin, His |
|
|
|
|
|
Красный |
Отрицательно |
Asp, Glu |
|
|
заряженные |
|
|
Синий |
Положительно |
Lys, Arg |
|
заряженные |
|||
|
|
Пример использования цветовой палитры (насколько это возможно в чёрно-белом представлении) представлен на рисунке 21.
Рисунок 21 – Представление множественного выравнивания белковых последовательностей
77

Важным элементом визуализации результатов множественного выравнивания являются аннотации или консенсусы.
Простейший вид аннотации использует программа ClustalW, в которой степень консервативности физико-химических свойств аминокислот в данной позиции выравнивания (в данном столбце) обозначается символами: "*" – "идентичность", ":" – "консервативность" и "." – "полуконсервативность" замен аминокислот в пределах данного столбца (рисунок 22).
Рисунок 22 – Результат множественного выравнивания аминокислотных последовательностей панкреатических эндонуклеаз лошади, малого полосатика и большого рыжего кенгуру
Более информативным способом визуализации результатов множественного выравнивания является генерация компьютером консенсусной строки, в которой изображаются те аминокислоты, которые наиболее часто встречаются в соответствующих столбцах данного выравнивания. Пример такого консенсуса – нижняя строка ("Consensus") на рисунке 21.
Для того, чтобы отобразить величину консервативности консенсусную строку изображают в виде гистограммы ("профиля") (рисунок 23(а)), в которой часто подписывают наиболее вероятные аминокислоты, причём размер символа соответствующей аминокислоты пропорционален частоте появления данной аминокислоты в данном столбце множественного выравнивания (рисунок 23(б, в)).
78
Рисунок 23 – Примеры аннотаций: а – гистограмма консервативности; б – консенсусная строка с символами аминокислот; в – формат представления Logo
Чтобы множественное выравнивание было информативным, оно должно содержать разные по эволюционному расстоянию последовательности.
Если все последовательности (1) чересчур близкие, то информация, которую они несут, избыточно дублируется, и это выравнивание малоинформативно.
А если все последовательности (2) далеки друг от друга, то трудно будет построить аккуратное выравнивание (кроме тех белков, для которых известны структуры), и в таком случае достоверность результатов и сделанных на их основе выводов оказывается под вопросом.
В идеале множественное выравнивание должно содержать широкий набор (спектр) белков разного уровня сходства, включающий далеко отстоящие экземпляры среди множества близких гомологов.
79