Материал: Огурцов А. Н. Методы бииоинформационного анализа
4.2.МЕТОДЫ МНОЖЕСТВЕННОГО ВЫРАВНИВАНИЯ
Кнастоящему времени были разработаны разнообразные методы, с помощью которых множественное выравнивание последовательностей известных белков можно применять для идентификации родственных последовательностей при поиске в базах данных. Эти методы реализованы в виде программ, наиболее важными из которых являются программы "Профили" (Profiles), "ПСИ-БЛАСТ" (PSI-BLAST) и "Скрытые марковские модели" (Hidden Markov Models, HMM).
Профили. Как правило, белки с подобной функцией имеют в своей структуре общий идентичный мотив. Поэтому предсказание мотивов
намного полезнее, чем поиск глобального подобия первичных последовательностей белков. Белки, обладающие подобными или сравнимыми функциями, весьма вероятно, являются производными формами общего белка-предка. Очень часто их последовательности (особенно мотивы) показывают значительное подобие. Множественное выравнивание последовательностей обычно позволяет обнаружить семейства родственных белков. Такой вид множественного выравнивания называют профилем.
Профиль (или весовая матрица) – это позиционная таблица счетов, в которую сведена информация о полном выравнивании последовательностей. Профили показывают, какие остатки могут находиться в данных позициях; какие позиции консервативны, а какие вырождены; которые позиции, или области, допускают вставки. В дополнение к данным, полученным из выравнивания, система счётов может включать в себя эволюционные веса и результаты анализа структур.
В литературе практически равновероятно используются термины профиль, паттерн, Position Weight Matrix (PWM, позиционная матрица счетов) и Position-Specific Weight Matrix (PSWM, позиционно-специфи- ческая матрица замен , или Position-Specific Scoring Matrix, PSSM).
Профиль выявляет регулярные комбинации, присутствующие во множественном выравнивании гомологичных последовательностей. Эти комбинации имеют большое значение.
80
•Они позволяют более аккуратно строить выравнивания дальнеродственных последовательностей.
•Наборы остатков высокой степени консервативности позволяют предположить их принадлежность к активному сайту и определять функцию.
•Консервативные паттерны облегчают идентификацию других похожих последовательностей.
•Консервативные паттерны могут использоваться для классификации подсемейств в множестве гомологов.
•Наборы остатков, в которых консервативность проявлена в низкой степени и в которых встречаются вставки и делеции, с высокой долей вероятности проявляются в петлях на поверхности белков. Эта информация применялась при разработке вакцин, поскольку указанные области с высокой долей вероятности стимулируют образование антител, которые будут хорошо взаимодействовать с нативными структурами.
•Методы предсказания структуры, основанные на множественном выравнивании последовательностей, являются более надежными, чем методы, построенные на анализе единственной последовательности. Например, моделирование гомологии кардинально зависит от построения правильного выравнивания последовательностей.
Для использования профилей в процессе идентификации гомологов, необходимо сопоставлять данную последовательность с последовательностями из базы данных, приведенными в таблице выравниваний, придавая консервативным позициям бόльший вес по сравнению с вариабельными позициями. Если известно, что какой-либо участок белковой цепи абсолютно консервативен, то процедура должна требовать обязательного наличия такого мотива.
В то же время слишком высокая степень жёсткости данной операции может повлечь за собой пропуск интересных дальних родственников, поэтому должны быть разрешены некоторые отклонения.
81
Вкачестве количественной меры консервативности каждой позиции
втаблице выровненных последовательностей сопоставляется вероятностное распределение аминокислот.
Например, для выравнивания
|
F |
K |
L |
L |
S |
H |
C |
L |
L |
V |
|
F |
K |
A |
F |
G |
Q |
T |
M |
F |
Q |
|
Y |
P |
I |
V |
G |
Q |
E |
L |
L |
G |
|
F |
P |
V |
V |
K |
E |
A |
I |
L |
K |
|
F |
K |
V |
L |
A |
A |
V |
I |
A |
D |
|
L |
E |
F |
I |
S |
E |
C |
I |
I |
Q |
|
F |
K |
L |
L |
G |
N |
V |
L |
V |
C |
можно составить весовую матрицу |
|
|
|
|
|
|||||
A |
-18 -10 -1 |
-8 8 -3 |
3 -10 -2 -8 |
|||||||
C |
-22 -33 -18 |
-18 -22 -26 |
22 |
-24 -19 |
-7 |
|||||
D |
-35 |
0 |
-32 |
-33 |
-7 |
6 |
-17 -34 -31 |
0 |
||
E |
-27 |
15 |
-25 |
-26 |
-9 |
23 |
-9 -24 -23 |
-1 |
||
F |
60 |
-30 |
12 |
14 |
-26 -29 -15 |
4 |
12 -29 |
|||
G |
-30 -20 -28 |
-32 |
28 |
-14 -23 -33 -27 |
-5 |
|||||
H |
-13 -12 -25 |
-25 -16 |
14 |
-22 -22 -23 -10 |
||||||
I |
3 |
-27 |
21 |
25 |
-29 -23 |
-8 |
33 |
19 -23 |
||
K |
-26 |
25 |
-25 |
-27 |
-6 |
4 |
-15 -27 -26 |
0 |
||
L |
14 |
-28 |
19 |
27 |
-27 -20 |
-9 |
33 |
26 -21 |
||
M |
3 |
-15 |
10 |
14 |
-17 -10 |
-9 |
25 |
12 -11 |
||
N |
-22 -6 -24 -27 1 |
8 -15 -24 -24 -4 |
||||||||
P |
-30 |
24 |
-26 |
-28 -14 -10 -22 -24 -26 -18 |
||||||
Q |
-32 |
5 |
-25 |
-26 |
-9 |
24 |
-16 -17 -23 |
7 |
||
R |
-18 |
9 |
-22 |
-22 -10 |
0 |
-18 -23 -22 |
-4 |
|||
S |
-22 -8 -16 -21 11 |
2 |
-1 -24 -19 -4 |
|||||||
T |
-10 -10 -6 |
-7 -5 -8 |
2 -10 -7 -11 |
|||||||
V |
0 |
-25 |
22 |
25 |
-19 -26 |
6 |
19 |
16 |
-16 |
|
W |
9 |
-25 -18 |
-19 -25 -27 -34 -20 -17 -28 |
|||||||
Y |
34 |
-18 |
-1 |
1 |
-23 -12 -19 |
0 |
0 -18 |
|||
элементы которой показывают вес данной аминокислоты для каждого столбца данного выравнивания.
При построении такой матрицы используется несколько методов. Во-первых репрезентативность профиля возрастает с увеличением
числа последовательностей, по которым производится выравнивание.
82
Во-вторых, учитывают вес замены данной аминокислоты i любой другой из наблюдаемых аминокислот, используя матрицы замен аминокислот, например, PAM250 или BLOSUM62. В таком случае аминокислота i может получить значение
ai = a1D(i,1) +a2D(i,2) + +a3D(i,20) .
Эта схема распределяет значения среди наблюдаемых аминокислот, взвешенных согласно вероятностям замен. Аминокислота в последовательности запроса получает большее значение, либо если она часто появляется в запросе в данной позиции, либо если для нее существует высокая вероятность появления с помощью мутации из остатков тех типов, которые обычны для данной позиции.
Данный подход более эффективен для распознавания отдалённых родственников при использовании ограниченного набора известных последовательностей. В этом случае вектор значений для аминокислот является произведением матрицы замен и вектора частот встречаемости остатков. Еще более правильным в данном случае было бы использование комбинации наблюдаемой выборки и частот встречаемости аминокислот в качестве распределения аминокислот.
Алгоритм, который реализует поиск оптимального выравнивания запрошенной последовательности и профиля среди всех возможных выравниваний является обобщением метода динамического программирования выравнивания двух последовательностей.
Недостаток простого профиля заключается в том, что множественное выравнивание последовательностей должно быть произведено заранее и учитывается в фиксированном виде.
Альтернативой является использование программы PSI-BLAST и скрытых марковских моделей (НММ), вычислительная (и прогностическая) мощность которых увеличивается с ростом объёма баз данных.
PSI-BLAST. PSI-BLAST – это программа, которая подбирает данные для последовательностей, аналогичных запрошенной. Она является обобщённой версией программы BLAST.
83
Программа BLAST (и её варианты) независимо сравнивает каждую запись в базе данных с запрошенной последовательностью.
Первый шаг программы BLAST – это поиск слов-кортежей некоторой установленной длины W со счётом выше некоторого порога Т. Величина W обычно равна 3 для последовательностей белка и 11 для последовательностей нуклеиновых кислот. Вначале BLAST выбирает слово-кортеж из последовательности запроса и продолжает удлинять его в обоих направлениях, сопоставляя с целевой последовательностью и одновременно подсчитывая счета совпадений и несовпадений, а также штрафы за введение и продолжение пропусков. Продолжение слова производится до тех пор, пока не будет достигнут некоторый предел S. BLAST продолжает отдельные пары совпадающих слов до тех пор, пока полный счёт выравнивания не снижается от максимальной величины до некоторого порога; в качестве результата программа выдает пары сегментов с высоким счётом.
BLAST – это эвристический алгоритм поиска, реализованный в различных программах пакета.
BLASTP сравнивает аминокислотную последовательность запроса с предметными последовательностями из базы данных белка.
BLASTN сравнивает запрашиваемую последовательность с предметными из базы данных нуклеотидных последовательностей.
BLASTX сравнивает результаты машинной смысловой трансляции
с шестью рамками считывания обеих нитей ДНК (Six-Frame Translation
или 6-Frame Translation) последовательности запроса нуклеотидов с содержимым базы данных последовательностей белков.
TBLASTN сравнивает белковую последовательность запроса с последовательностями из базы данных нуклеотидных последовательностей, динамически транслируемых с шестью рамками считывания (обе нити).
TBLASTX сравнивает продукты трансляции с шестью рамками считывания нуклеотидной последовательности запроса с продуктами трансляции с шестью рамками считывания последовательностей из базы данных последовательностей нуклеотидов.
84
PSI-BLAST сравнивает аминокислотную последовательность запроса с предметными последовательностями из белковых баз данных.
Программа PSI-BLAST начинает работу с поэлементного сравнения каждой записи в базе данных с запрошенной последовательностью запроса. Затем она строит локальное множественное выравнивание последовательностей, полученных при первичном запросе, и затем обращается к базе данных, используя уже это множественное выравнивание.
Затем процесс повторяется (по полученному набору кандидатов снова строится множественное выравнивание), и результат уточняется в ходе нескольких итераций, пока не исчерпается заданное количество циклов или пока процедура не сойдется, т. е. пока результаты двух последовательных запросов не совпадут.
Причиной, по которой потребовалось создание программы BLAST, являлось то, что полномасштабные методы динамического программирования недостаточно быстры для задачи полного поиска в большой базе данных. Часто база данных содержит последовательности, очень похожие на запрошенную последовательность. Менее точные, но более быстрые программы вполне способны идентифицировать близкие совпадения, что в большинстве случаев и требуется.
Например, если существует необходимость найти гомологи мышиного белка в геноме человека, то степень сходства скорее всего будет высокой, и более быстрые методы подойдут для решения этой задачи.
Но в случае необходимости нахождения гомологов человеческого белка в С. elegans или дрожжах, различия будут более тонкими, и, следовательно, требуется программа с более высокой степенью точности.
Метод, на котором основана программа BLAST, вообще говоря, подобен анализу точечных матриц совпадений, которые выявляют хорошо совпадающие локальные участки. Для каждой записи в базе данных проверяются короткие сопредельные участки, совпадающие с короткими сопредельными участками запрошенной последовательности, для чего используется матрица аминокислотных замен, но без пропусков.
85
Участки фиксированной длины быстро определяются при использовании
таблиц поиска (хеш-таблиц).
Таблицей поиска (Lookup table) называют – структуру данных, обычно массив или ассоциативный массив, используемая с целью
заменить вычисления на операцию простого поиска. Увеличение скорости может достигается за счёт того, что провести простой поиск в памяти компьютера требует гораздо меньше машинного времени, чем выполнить громоздкие вычисления. Примеры таблиц поиска – это таблицы тригонометрических функций или таблицы логарифмов (например, таблицы Брадиса), которые были широко распространены до начала массового производства инженерных калькуляторов.
Как только программа BLAST определяет подходящий участок, она пытается его расширить. В некоторых версиях программы допускается наличие пропусков. На выходе программа предлагает набор локальных сегментных совпадений.
Программа PSI-BLAST, которая использует итерационный поиск последовательности, гораздо эффективнее BLAST при изучении менее близкородственных связей. PSI-BLAST точно определяет в три раза больше гомологов, чем BLAST, в фрагментах, в которых совпадения составляют меньше 30%. Следовательно, этот метод весьма хорошо применим в случае анализа целых геномов. PSI-BLAST способна идентифицировать белковые домены известной структуры для 39% генов
M.genitaliurn, 24% генов дрожжей и 21% генов С. elegans.
Единственным более эффективным методом, основанным на анализе последовательностей, является метод скрытых марковских моделей.
4.3. СКРЫТЫЕ МАРКОВСКИЕ МОДЕЛИ
Скрытая марковская модель (Hidden Markov model, HMM) – это статистическая модель, имитирующая работу процесса, похожего на марковский процесс с неизвестными параметрами, в которой задачей ставится разгадывание (восстановление) неизвестных параметров на основе наблюдаемых. Полученные параметры могут быть использованы в
86
дальнейшем анализе, например, для распознавания образов. Первые заметки о скрытых марковских моделях опубликовал Баум в 1960-х, и уже в 70-х их впервые применили при распознавании речи. С середины 1980-х СММ применяются при анализе биологических последовательностей, в частности, ДНК.
Марковский процесс – это случайный процесс, эволюция которого после любого заданного значения временного параметра t не зависит от эволюции, предшествовавшей t , при условии, что значение процесса в этот момент фиксировано ("будущее" процесса не зависит от "прошлого" при известном "настоящем"; или по определению Вентцеля: "будущее" процесса зависит от "прошлого" лишь через "настоящее").
Определяющее марковский процесс свойство принято называть марковским; впервые оно было сформулировано А.А. Марковым. Марков, Андрей Андреевич (1856–1922) – выдающийся русский математик, внёсший большой вклад в теорию вероятностей, математический анализ и теорию чисел.
Вработах 1907 г. А.А. Марков положил начало изучению последовательностей зависимых испытаний и связанных с ними сумм случайных величин. Это направление исследований известно под названием теории цепей Маркова (Markov Chain). Кроме биоинформатики скрытые марковские модели применяются в криптоанализе и машинном переводе.
Вбиоинформатике скрытые марковские модели служат для описания тонких различий, существующих между семействами гомологичных последовательностей. Метод скрытых марковских моделей (Hidden Markov model, HMM) эффективен при сравнении дальних родственников и при предсказании путей сворачивания белков. Только этот метод, полностью базирующийся на анализе последовательностей (т. е. не использующий структурную информацию), может соперничать с программой PSI-BLAST при идентификации дальних гомологов.
Хотя НММ работает с множественными выравниваниями последовательностей, но НММ также часто используется для генерации последовательностей. Обычная таблица множественного выравнивания последовательностей, вообще говоря, также может использоваться для
87
генерации этих последовательностей путём выборки аминокислот в последовательных позициях. Каждая аминокислота выбирается из вероятностного позиционно-специфического распределения, полученного из профиля.
Модели НММ являются более общими методами, чем профили, поскольку включают вероятность возникновения делеций или вставок в генерируемой последовательности с позиционно-зависимыми штрафами для них.
Если применение профилей требует предварительного построения множественного выравнивания последовательностей и при таком подходе статистика запросов определяется после построения выравнивания, то НММ одновременно выполняет и выравнивание, и оценку вероятностей.
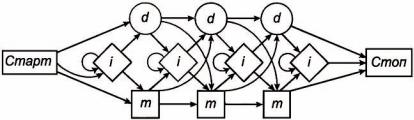
Рисунок 24 схематично иллюстрирует механизм генерации последовательностей. На рисунке показаны три позиции и набор состояний (m), (i), (d).
Рисунок 24 – Структура скрытой марковской модели
Начиная со "Старт" НММ перемещается по стрелкам до "Стоп". Каждая стрелка приводит к определённому состоянию (state) системы. В каждом из этих состояний предпринимается (1) определенное действие (например, задаётся аминокислота) и затем (2) выбирается стрелка, которая переводит в следующее состояние системы. Действие и выбор следующего состояния управляются определёнными наборами вероятностей.
На каждой позиции, на которой вводится аминокислотный остаток, один набор вероятностей задан для процесса выбора одной из 20
88
стандартных аминокислот, а второй набор вероятностей – для процесса выбора пути перехода к следующей стадии процесса. Оба этих набора вероятностей подбираются так, чтобы задать адекватное описание определенного семейства последовательностей. Используя такой подход, и варьируя только позиционно-специфические таблицы вероятностей, одинаковый алгоритм может быть использован для различных семейств последовательностей. На рисунке 24 каждой из трёх позиций во множественном выравнивании НММ соответствуют сопоставление (m) (mach) и делеция (d). Вставки (i) могут быть как между позициями остатков, так и в начале, и в конце выравнивания.
Если данная позиция соответствует сопоставлению (m), то в выравнивание вставляется соответствующий аминокислотный остаток.
Вданном случае слово "сопоставление" означает только то, что некоторые аминокислоты есть и в НММ-последовательности, и в итоговой последовательности, а не то, что это должны быть обязательно одинаковые аминокислоты.
Вероятность выбора одной из 20 аминокислот в данной позиции сопоставления определяется параметрами используемой модели, то есть позиционно-специфичными вероятностями вставки аминокислот в данной позиции.
Вслучае делеции (d) данная колонка множественного выравнивания пропускается. Введение первой делеции означает открытие интервала делеции (gap-open), а вероятность такого введения определяет величину штрафа за введение делеции, которая также является позиционноспецифичной. Введение второй делеции сразу вслед за первой означает продолжение делеции (gap-extension), за что также может назначаться штраф (см. п. 2.2).
Вставки (i) помещают между двумя последовательными позициями
ввыравнивании. Если программа вводит вставку, то в генерируемую последовательность вставляется остаток, не совпадающий (в данной позиции) с остатками в последовательностях, по отношению к которым производится множественное выравнивание. За первой вставкой может следовать вторая и т. д., образуя вставку размером больше, чем один остаток.
89