Материал: Біологічна та біоорганічна хімія_Мардашко О.О._ изд. 2008-342 с._ОНМедУ-2012
піруваткарбоксилази, що каталізує перетворення пірувату на оксалоацетат.
2)Коферменти — фосфорні ефіри вітамінів.
До них належать тіамінпірофосфат (ТПФ), піридоксальфосфат (ПАЛФ), піридоксамінфосфат
(ПАМФ). Тіамінпірофосфат входить до складу таких ферментів, як піруват та α -кетоглутарат- дегідрогеназний комплекси, транскетолаза. Коферменти ПАЛФ і ПАМФ є фосфорними ефірами
вітаміну В6, вони входять до складу амінотрансфераз, що каталізують оборотне перетворення амінокислот на кетокислоти. Піридоксальфосфат
єкоферментом декарбоксилаз, що каталізують утворення біогенних амінів з амінокислот.
3)Коферменти-нуклеотиди — ця група ко-
ферментів поділяється на дві підгрупи: кофермен- ти-мононуклеотиди і коферменти-динуклеотиди.
а) До коферментів-мононуклеотидів належать АТФ, ГТФ, ЦТФ, УТФ і ФМН. Як кофермент АТФ входить до складу таких ферментів, як гексокіназа і гліцеролкіназа. Перший каталізує перенос фосфату від АТФ на гексози з утворенням фосфорних ефірів гексоз. Гліцеролкіназа каталізує перенос фосфату з АТФ на гліцерол з утворенням активної форми гліцерол-3-фосфату. Так само АТФ є коферментом ферментів аміноацилсинтетаз, що каталізують перенос АМФ від АТФ на амінокислоту з утворенням аміноациладенілатів — активної форми амінокислоти, необхідної в процесі біосинтезу білка. Також АТФ є коферментом ферментів, які каталізують перенос аденозину з АТФ на метіонін з утворенням активної форми метіоніну — S-аденозилметіоніну; ГТФ бере участь у біосинтезі білків, ЦТФ — у біосинтезі фосфоліпідів, УТФ — у біосинтезі глікогену; ФМН є коферментом оксидази альфа-аміно- кислот і НАДН-дегідрогенази, cкладається з pибoфлaвіну і залишку фocфopнoї киcлoти.
б) Коферменти-динуклеотиди. До них належать НАД+, НАДФ+, ФАД і коензим А. Одним із мононуклеотидів усіх цих коферментів є аденозинмонофосфат. Другою частиною цих коферментів є специфічний нуклеотид, у якому азотиста основа представлена відповідним вітаміном.
Кoфepмeнт HAД+ — нікотинамідаденіндинyклeoтид — складається з двоx мoнoнyклeoтидів: пepший — aдeнілoвa киcлoтa, другий — aмід нікoтинoвoї киcлoти, pибoзa та фocфaт. Обидва мoнoнyклeoтиди з’єднані між собою залишками фocфорної кислоти. Нікoтинaмідaдeніндинyклeoтидфocфaт (HAДФ+) тaкож є динyклeoтидoм, але містить у cвoєму складі нe двa, a тpи залишки фocфopної кислоти.
Коферменти НАД+ і НАДФ+ здатні оборотно приймати електрони і протони, тому вони входять до складу дегідрогеназ. Окиснені форми коферментів у реакціях позначають НАД+, НАДФ+, а відновлені — НАДН+Н+ і НАДФН+Н+. Усі фepмeнти, що містять як кoфepмeнт HAД+ чи НAДФ+, звутьcя нікoтинaмідними (піpидинoвими) фepмeнтaми, їx відомо більше 150. Taк,
HAД+ вxoдить до складу тaкиx фepмeнтів, як піpyвaтдeгідpoгeнaзa, α -кeтoглyтapaтдeгідpo- гeнaзa, лактaтдeгідpoгeнaзa, β -гідроксіацил-
KoA-дeгідpoгeнaзa, гліцеральдегідфосфатдегідрогеназа та ін.; НАДФ+ як кoфepмeнт вxoдить до складу глюкoзo-6-фocфaтдeгідpoгeнaзи, ізoцитpaтдeгідpoгeнaзи та ін.
Флaвінaдeніндинyклeoтид (ФAД) складається з aдeнілoвoї киcлoти та ФМН (флaвінмoнoнyклeoтиду), які з’єднані між coбoю піpoфocфaтним з’язком. Ферменти, до складу яких вxoдить ФAД чи ФМН, нaзивaютьcя флaвінoвими фepмeнтами (флaвoпpотеїнами). Особливістю коферментів ФМН і ФАД є здатність до реакцій окис- нення-відновлення. Найважливішими ферментами, що містять кофермент ФАД, є піруват та α - кетоглутаратдегідрогеназний комплекси, ацил- КоА-дегідрогеназа, сукцинатдегідрогеназа.
Koензим A, чи кoфepмeнт A (KoA), складається з aдeнілoвoї киcлoти і пaнтoтeїну, які з’єднані між coбoю піpoфocфaтним зв’язком, пaнтoтeїн — з вітaміну пaнтoтeнoвoї киcлoти, мepкaптоетилaміну та залишку фocфopнoї киcлoти. Коензим А є коферментом у піруват і α -кетоглутарат- дегідрогеназному комплексах, бере участь в утворенні активних форм жирних кислот — ацилКоА.
4) Коферменти — металовмісні комплекси. До них належать ферумпорфіринові комплекси, інші металовмісні коферменти. Головною складовою частиною ферумпорфіринових комплексів є 4 пірольних кільця, зв’язані із Ферумом. Ферумпорфіринові комплекси входять до складу таких ферментів, як цитохроми, каталаза, пероксидаза. Цитохроми — ферменти тканинного дихання, що каталізують передачу електронів на кисень. Каталаза перетворює пероксид гідрогену на воду та кисень. Пероксидаза також каталізує перетворення перекису гідрогену за участю атомів Гідрогену. До металовмісних ферментів належать ксантиноксидаза (молібденфлавопротеїн, що каталізує окиснення ксантину до сечової кислоти), церулоплазмін і цитохромоксидаза (купрумвмісні протеїни) каталізують реакції окиснення, карбоангідраза — цинковмісний фермент та ін.
6.3. МЕХАНІЗМ ДІЇ ТА ВИЗНАЧЕННЯ АКТИВНОСТІ ФЕРМЕНТІВ. КІНЕТИКА
Швидкість хімічних реакцій залежить: а) від концентрації реагуючих речовин; б) від ступеня хімічної спорідненості реагуючих речовин; в) від кількості активних молекул реагуючих речовин. Якщо у лабораторних умовах можна змінювати концентрацію сполук, підбирати сполуки, які вступають до хімічної реакції, збільшувати кількість активних молекул (наприклад нагріванням), то в біологічних системах, зокрема в клітинах, всі перелічені умови має виконати фермент, що бере участь у реакції. Молекули субстрату, що знаходяться в середовищі у певній концентрації, адсорбуються на поверхні ферменту, особливо біля його активного центру, при цьому концентрація молекул субстрату на фер-
89
менті багаторазово збільшується. У формуванні ферментсубстратного комплексу беруть участь водневі зв’язки, електростатичні та гідрофобні взаємодії, ковалентні та координаційні зв’язки. Індукована відповідність субстрату та ферменту створюється зміною конформації білкової молекули, геометричною й електронно-топографічною перебудовою молекули субстрату.
Ферменти прискорюють реакції за рахунок зниження енергії активації, яка необхідна для переходу молекул речовин у активний стан. Якщо для перебігу хімічної реакції між сполуками А і В потрібно витратити певну кількість енергії Е, то утворення ферментсубстратного комплексу потребує енергії Е1, а сполучення А і В за участю ферменту потребує енергії Е2, що сумарно значно менше, ніж Е.
А+ В →Е АВ
А+ Ф Е→ 1 АФ
АФ + В →Е2 АВ + Ф Е1 + Е2 < Е
Так, каталаза знижує енергію активації з 77,8 до 22,5 кДж/моль, тобто більш ніж утричі.
Основною умовою ферментативної реакції є те, що для здійснення свого впливу фермент повинен з’єднатися з субстратом. Після утворення ферментсубстратного комплексу субстрат перетворюється на продукт реакції та вивільняється, а фермент повертається у вихідний стан. Швидкість реакції вимірюється кількістю продукту, що утворюється під впливом ферменту (або кількістю субстрату, що прореагував) за одиницю часу. Швидкість ферментативних реакцій залежить від впливу зовнішніх умов (температура, рН середовища, вплив природних і чужорідних сполук тощо).
Основи кінетики ферментативних реакцій були закладені в роботах Міхаеліса і Ментен (1913). Швидкість ферментативної реакції є мірою каталітичної активності ферменту (позначається просто — активність ферменту).
Шляхи визначення ферментативної активності: а) за кількістю субстрату, перетвореного за одиницю часу (пепсин, трипсин); б) за кількістю продукту реакції, утвореного за одиницю часу (ліпаза); в) за часом, необхідним для перетворення певної кількості субстрату (амілаза).
Залежність швидкості каталізу від концентрації субстрату й ферменту
Етапи реакцій:
а) взаємодія ферменту (Е) і субстрату (S), утворення проміжного ферментсубстратного комплексу (ЕS). Слід зазначити, що кожна хімічна реакція має свою константу швидкості (k+1). Але не всі 100 % молекул ферментсубстратного комплексу, що утворилися, візьмуть участь у подальших перетвореннях. Певна частина із них дисоціює з константою швидкості, характерною для кожної реакції (k–1). Кількість утворених молекул ферментсубстратного комплексу залежатиме від співвідношення (k+1)/(k–1);
б) перетворення ферментсубстратного комплексу і перебіг хімічної реакції відбуватиметься також із певною швидкістю, характерною для кожної реакції (k+2);
в) дисоціація комплексу і відділення продуктів реакції від ферменту. Таким чином, кожна реакція має певне співвідношення між константами швидкостей «прямої» реакції утворення ферментсубстратного комплексу, «зворотної» реакції його дисоціації та реакції розпаду на фермент і продукт реакції (рівняння Міхаеліса — Ментен):
|
k+1 |
|
k+2 |
|
|
|||
E + S |
|
|
|
ES |
|
... |
|
E + P |
|
|
|
|
|||||
|
|
|||||||
k-1 |
|
|||||||
|
|
|
|
|
|
|
||
k-1 + k-1 , Кm = ————
k-1
де Кm — константа Міхаеліса (концентрація субстрату, при якій швидкість реакції дорівнює половині від максимальної).
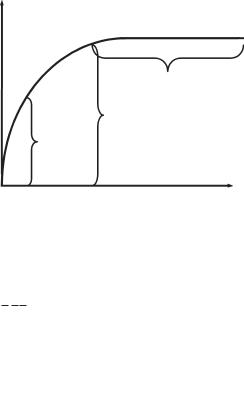
Графічне зображення рівняння Міхаеліса — Ментен показує залежність швидкості реакції від концентрації субстрату (рис. 6.2):
V
Vmax
Реакція нульового порядку, швидкість не залежить від [S]
1/2Vmax Реакція першого порядку, швидкість залежить від [S]
Кm |
[S] |
Рис. 6.2. Залежність швидкості реакції від концентрації субстрату
Швидкість реакції не пропорційна концент- |
Чим активніший фермент, тим нижче значен- |
рації субстрату. При збільшенні концентрації |
|
субстрату швидкість спочатку збільшується, а |
ня його Km. По тому, як змінюється швидкість ре- |
потім наближається до деякої постійної величи- |
акції при різних концентраціях субстрату, мож- |
ни, а крива зміни швидкості — до певного гра- |
на судити про порядок реакції, який необхідно |
ничного значення, що відповідає максимальній |
знати для роботи з ферментами й для правильно- |
швидкості. |
го визначення їхньої активності в клінічних ла- |
90
бораторіях. Порядок реакції може варіювати від |
Збільшення кількості молекул ферменту, що |
|
нульового й вище. При нульовому порядку |
досягається шляхом природної стимуляції їхньо- |
|
швидкість реакції — величина постійна, не зале- |
го утворення або за допомогою препаратів, доз- |
|
жить від концентрації субстрату. При цьому |
воляє або відновлювати порушену швидкість ре- |
|
швидкість реакції максимальна (Vmax). |
акцій, або пристосувати необхідні біохімічні ре- |
|
При першому порядку реакції її швидкість |
акції до нових умов життєдіяльності. |
|
прямо пропорційна концентрації одного з суб- |
|
|
стратів. Для того, щоб правильно визначити ак- |
|
|
тивність ферменту, потрібно домогтися нульово- |
Залежність швидкості ферментативної |
|
го порядку реакції, тобто визначити швидкість |
реакції від рН середовища |
|
ферментативної реакції при насичуючих концен- |
Ферментативні реакції дуже чутливі до рН се- |
|
траціях субстрату. У цьому випадку всі зміни |
||
редовища. Це пов’язане з трьома факторами: |
||
швидкості реакції залежатимуть тільки від кіль- |
||
кості ферменту. |
1) при екстремальних значеннях рН необорот- |
|
но змінюється структура ферменту, оскільки в цих |
||
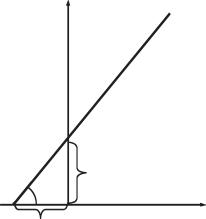
Застосовуючи метод подвійних зворотних ве- |
||
личин, Г. Лайнуївер і Д. Берк перетворили гіпер- |
умовах може відбуватися денатурація білка, а в |
|
болічну криву рівняння Міхаеліса — Ментен на |
деяких випадках — і зміна характеру зв’язку між |
|
графік лінійної залежності (рис. 6.3): |
апоферментом і коферментом; |
|
2) зміна рН у більшості випадків порушує ха- |
||
y = ax + b, |
||
рактер іонізації субстрату; |
||
де у = 1/V; х = 1/[S], а тангенс кута нахилу дорів- |
3) значення рН впливає на іонізацію кислих |
|
і основних груп амінокислотних залишків актив- |
||
нює Кm/Vmax. |
ного центру, які беруть участь у зв’язуванні суб- |
|
|
||
|
страту (у контактній ділянці) або в його перетво- |
|
|
ренні (у каталітичній ділянці). |
|
|
Тому специфічний вплив рН може спричини- |
|
|
ти: зміну спорідненості субстрату до ферменту; |
|
|
зміну каталітичної активності ферменту; обидва |
|
|
порушення разом. Існує залежність швидкості |
|
|
ферментативної реакції від рН середовища, для |
|
|
кожного ферменту існує свій оптимум рН, при |
|
|
якому швидкість реакції, що він каталізує, мак- |
|
|
симальна. Відхилення рН у ту чи іншу сторону |
|
|
веде до зниження швидкості ферментативної ре- |
|
|
акції. Більша частина ферментів клітин має оп- |
|
|
тимум рН, близький до нейтрального, тобто |
|
α |
співпадаючий з фізіологічними значеннями рН. |
|
|
Діапазон коливань рН у фізіологічних умовах |
|
1/[S] |
незначний, але зміни рН на обмеженій ділянці |
|
–1/Кm |
клітини можуть бути. Вони впливають на діяль- |
|
Рис. 6.3. Графік рівняння Лайнуївера — Берка |
ність ферментів. Наприклад, при активній м’я- |
|
зовій роботі накопичується молочна кислота, яка |
||
|
||
|
зрушує рН середовища у м’язових клітинах у |
|
Щоб оцінити умови роботи будь-якого фермен- |
кислу сторону, що змінює швидкість фермента- |
|
ту в клітинах організму, необхідно знати, як ре- |
тивних реакцій. Знання оптимумів рН окремих |
|
ально змінюються в них концентрації субстратів. |
ферментів важливе для практичної медицини. |
|
У фізіологічних умовах ферменти майже ніко- |
Наприклад, пепсин для активного гідролізу |
|
ли не працюють на повну силу, тому що концен- |
білків у шлунку потребує сильнокислого середо- |
|
трації субстратів для них далекі від насичуючих. |
вища, тому для відновлення порушеної актив- |
|
Можливо, що тільки єдиний субстрат, необхід- |
ності ендогенного пепсину необхідно застосову- |
|
ний для дії гідролаз, — вода — присутня в кліти- |
вати кислі речовини. Препарат пепсину вжива- |
|
нах у насичуючих концентраціях, за винятком |
ють із соляною кислотою, що створює потрібний |
|
випадків, коли структурна локалізація фермен- |
рН. |
|
ту обмежує доступ до активного центру. |
Залежність швидкості ферментативної |
|
Залежність швидкості реакції від кількості фер- |
||
реакції від температури |
||
менту має лінійний характер, що відрізняє фер- |
||
мент від небіологічних каталізаторів. Із цього |
Оптимальні значення температури для біль- |
|
треба зробити висновок, що чим більше молекул |
||
шості ферментів перебувають у межах 20–40 °С. |
||
даного ферменту в клітині організму порівняно |
Термолабільність ферментів пов’язана з їхньою |
|
з іншими, тим вища в ній швидкість реакції, що |
білковою будовою. Деякі ферменти денатурують |
|
каталізує цей фермент. Якщо ж якого-небудь фер- |
уже при температурі близько 40 °С, але основна |
|
менту недостатньо (порушений синтез), то |
частина їх інактивується при температурі вище |
|
швидкість реакції, що він каталізує, обмежує хід |
40–50 °С. У окремих ферментів при температурах, |
|
пов’язаних із нею біохімічних процесів. |
близьких до 0 °С, настає денатурація. Однак де- |
|
|
91

які ферменти не підпорядковуються цим законо- |
му випадку їх називають активаторами) або |
||||||||
мірностям. Наприклад, фермент каталаза найак- |
вповільнюватися (у цьому випадку їх називають |
||||||||
тивніший при температурах, що наближаються |
інгібіторами). |
|
|
||||||
до 0 °С. Існують і термостабільні ферменти — |
|
|
|
|
|
|
|
|
|
так, аденілаткіназа витримує короткочасно тем- |
Активація ферментів |
|
|||||||
пературу 100 °С без інактивації. Мікроорганіз- |
Активація ферментів визначається за приско- |
||||||||
ми, що живуть у гарячих джерелах, містять білки, |
|||||||||
ренням біохімічних реакцій, що настає після дії |
|||||||||
у тому числі й ферменти, що відрізняються висо- |
|||||||||
кою термостабільністю. |
модифікатора. Одна група активаторів — це ре- |
||||||||
човини, що впливають на ділянку активного |
|||||||||
Штучне охолодження організму (гібернація) |
|||||||||
використовується в клініці для проведення хірур- |
центру ферменту. До них належать кофактори |
||||||||
гічних операцій. Охолодження тіла сповільнює |
ферментів і субстрати. Кофактори (іони металів і |
||||||||
швидкість ферментативних реакцій, що дозволяє |
коферменти) є не тільки обов’язковими структур- |
||||||||
знизити витрату речовин і довше зберегти жит- |
ними елементами складних ферментів, але й, по |
||||||||
тєздатність клітин організму. |
суті, їхніми активаторами. Для Na+, K+-АТФази, |
||||||||
що здійснює транспорт одновалентних катіонів |
|||||||||
Підвищення температури тіла (гарячковий |
|||||||||
стан — наприклад при інфекціях) прискорює |
через клітинну мембрану, необхідні як активато- |
||||||||
біохімічні реакції, що каталізують ферменти. |
ри іони Магнію, Натрію й Калію. |
||||||||
Збільшення температури тіла на кожний градус |
Активація за допомогою іонів металів здійс- |
||||||||
нюється за різними механізмами. У деяких фер- |
|||||||||
підвищує швидкість реакції приблизно на 20 %. |
|||||||||
При високих температурах близько 39–40 °С |
ментах вони входять до складу каталітичної |
||||||||
марне використання ендогенних субстратів у |
ділянки, іони металів полегшують зв’язування |
||||||||
клітинах хворого організму обов’язково потрібно |
субстрату з активним центром ферменту. Іноді |
||||||||
поповнювати їхнім надходженням з їжею або |
метал з’єднується не з ферментом, а з субстратом, |
||||||||
ліками. Крім того, при температурі близько 40 °С |
утворюючи металосубстратний комплекс. Іони |
||||||||
частина досить термолабільних ферментів може |
металів, коферменти, їхні попередники й активні |
||||||||
денатуруватися, що порушує природний хід біо- |
аналоги, субстрати можна використовувати на |
||||||||
хімічних процесів. |
практиці як препарати, що активують ферменти. |
||||||||
Активація деяких ферментів може здійснюва- |
|||||||||
|
|||||||||
Одиниці активності ферментів |
тися шляхом модифікації, що не зачіпає активний |
||||||||
центр їхніх молекул. Можливі кілька варіантів |
|||||||||
|
|||||||||
Для вираження концентрації ферменту й |
такої модифікації: |
|
|
||||||
кількісної оцінки його активності рекомендована |
1. Активація неактивного попередника — |
||||||||
стандартна міжнародна одиниця (Е або U). Оди- |
проферменту, або зимогену. |
||||||||
ниця активності ферменту — та кількість його, |
2. Активація шляхом приєднання якої-небудь |
||||||||
яка в оптимальних умовах каталізує перетворен- |
специфічної модифікуючої групи до молекули |
||||||||
ня 1 мікромоля субстрату або утворення 1 мікро- |
ферменту, хімічна модифікація (фосфорилування |
||||||||
моля продукту за 1 хв (мкмоль/хв). Існує також |
й дефосфорилування апоферменту). |
||||||||
одиниця каталітичної активності «катал», що |
3. Регуляція за принципом зворотного зв’яз- |
||||||||
являє собою кількість ферменту, здатну перетво- |
ку (ретроінгібування через алостеричний центр). |
||||||||
рити 1 моль субстрату за 1 c у стандартних умо- |
4. Конкуренція за субстрат або кофермент. |
||||||||
вах (температура, рН, насичуюча концентрація |
5. Активація шляхом дисоціації неактивного |
||||||||
субстрату). |
комплексу білок — активний фермент. |
||||||||
Рекомендовано вимірювати активність фер- |
|
|
|
|
|
|
|
|
|
менту при температурі 25 °С при оптимумі рН і |
|
|
|
|
|
|
|
|
|
концентрації субстрату, що перевищує концен- |
Інгібування ферментів |
|
|||||||
трацію насичення. У цих випадках швидкість |
Інгібітори характеризуються, насамперед, та- |
||||||||
реакції відповідає нульовому порядку реакції |
|||||||||
кою загальною ознакою, як міцність зв’язуван- |
|||||||||
відносно субстрату й залежатиме тільки від кон- |
|||||||||
центрації ферменту. У практичній роботі з фер- |
ня з ферментом. За цією ознакою інгібітори |
||||||||
ментами користуються поняттями питомої й мо- |
діляться на дві групи: оборотні й необоротні. За- |
||||||||
лярної активності. |
рахувати інгібітори до відповідної групи дозво- |
||||||||
ляє критерій відновлення активності ферменту |
|||||||||
|
|||||||||
|
після діалізу або сильного розведення розчину |
||||||||
6.4. РЕГУЛЯЦІЯ АКТИВНОСТІ |
ферменту з інгібітором. |
|
|||||||
Оборотне інгібування: |
|
||||||||
ФЕРМЕНТІВ. ІНГІБІТОРИ. |
Е + І |
|
|
|
|
ЕІ |
Е — фермент |
||
АКТИВАТОРИ |
|
|
|
|
|||||
|
|
|
|||||||
|
|
|
|
|
|
|
|
||
Регуляція активності ферментів може здійсню- |
Необоротне інгібування: |
||||||||
Е + І |
|
|
|
|
|
ЕІ |
І — інгібітор |
||
ватися шляхом взаємодії з ними різних біологіч- |
|
|
|||||||
|
|||||||||
них компонентів і чужорідних сполук (ліків і от- |
|
|
|
|
|
|
|
|
|
рут), які прийнято називати модифікаторами або |
Частіше відбувається оборотне інгібування, |
||||||||
регуляторами ферментів. Під дією модифікаторів |
що підлягає кількісному вивченню на основі |
||||||||
на фермент реакція може прискорюватися (у цьо- |
рівняння Міхаеліса — Ментен. Оборотне інгібу- |
||||||||
92
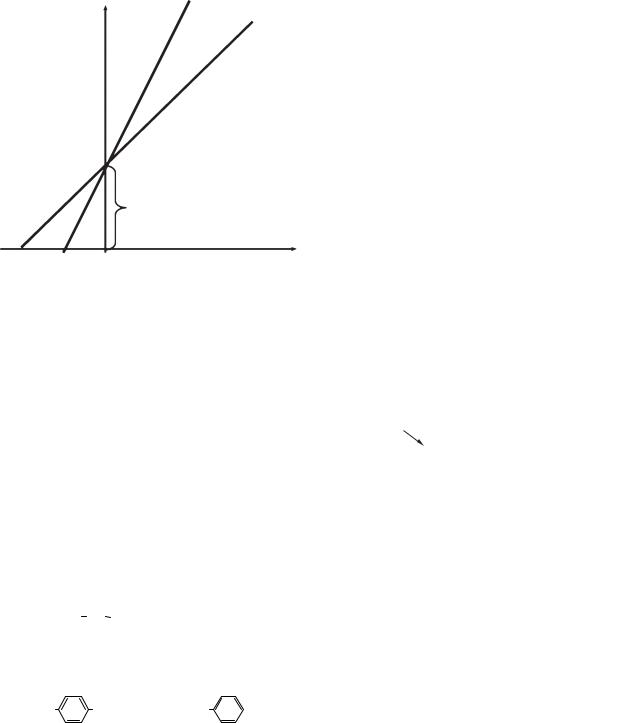
вання у свою чергу поділяють на конкурентне й неконкурентне — залежно від того, чи можливо відновити активність ферментативної реакції шляхом збільшення концентрації субстрату. Конкурентним інгібуванням називається гальмування ферментативної реакції, спричинене зв’язуванням інгібітора з активним центром ферменту, інгібітор і субстрат, будучи подібними за будовою, конкурують за активний центр ферменту. З активним центром зв’язується та сполука, молекул якої більше (рис. 6.4).
|
2 |
1/V |
1 |
-1/Km -1/Kmі |
1/[S] |
Рис. 6.4. Конкурентне інгібування ферментів: 1 — графік Лайнуївера — Берка без інгібітора;
2 — графік Лайнуївера — Берка в присутності конкурентного інгібітора
При конкурентному типі інгібування інгібітор збільшує значення Kmi, не впливаючи на макси-
мальну швидкість Vmax. Це означає, що при досить високій концентрації субстрату [S] інгібітор
витісняється молекулами субстрату з комплексу EI. Класичним прикладом подібного типу інгібування є вплив різних речовин на активність сукцинатдегідрогенази — ферменту циклу Кребса. Його природним субстратом є сукцинат, а подібним до нього конкурентним інгібітором — оксалоацетат, проміжний продукт того ж циклу Кребса.
Аналогічним конкурентним інгібітором сукцинатдегідрогенази є малонова кислота, що часто використовується в біохімічних дослідженнях.
O
-OOC |
|
СН2 СН2 СОО- |
-OOC |
|
|
|
|
|
|
СОО- |
|
|
С |
|
СН2 |
|
|||||
|
|
|
|
|||||||
Сукцинат Оксалоацетат
Метод конкурентного інгібування знайшов широке застосування в медичній практиці:
H2N |
COOH |
H2N |
|
|
SO2 |
|
NH2 |
|
|
|
|||||
n-Амінобензенова кислота |
|
Сульфаніламід |
|||||
Для лікування деяких інфекційних захворювань, викликаних бактеріями, застосовують сульфаніламідні препарати. Виявилося, що ці препарати мають структурну подібність із параамінобензойною кислотою, яку бактеріальна клітина використовує для синтезу фолієвої кислоти, що є складовою частиною ферментів бактерій. Завдяки структурній подібності сульфаніламід блокує дію ферменту шляхом витиснення параамінобензойної кислоти з комплексу з ферментом, що синтезує фолієву кислоту.
Неконкурентним інгібуванням ферменту називається гальмування, пов’язане з впливом інгібітора на каталітичне перетворення, але не на зв’язування субстрату з ферментом. Неконкурентний інгібітор може зв’язуватися безпосередньо з каталітичними групами активного центру ферменту або з ферментом поза активним центром, що заважає взаємодії з субстратом. Дія неконкурентних інгібіторів не усувається додаванням надлишку субстрату.
У присутності інгібіторів такого типу частина ферментів інактивується, швидкість реакції (зокрема максимальна швидкість) зменшується, але константа Міхаеліса залишається без змін.
Неконкурентне інгібування може бути оборотним і необоротним. При оборотному неконкурентному інгібуванні субстрат S й інгібітор I зв’я- зуються з різними центрами, тому з’являється можливість утворення як комплексу EI, так і комплексу EIS; останній може розпадатися зі звільненням продукту, але з меншою швидкістю, ніж комплекс ES (рис. 6.5).
1/V |
2 |
1 |
|
||
|
|
1/Vі
1/Vmax
-1/Km = -1/Km |
1/[S] |
і |
|
Рис. 6.5. Неконкурентне інгібування ферментів: 1 — графік Лайнуївера — Берка без інгібітора; 2 — графік Лайнуївера — Берка в присутності неконкурентного інгібітора
До неконкурентних інгібіторів належать іони важких металів (Меркурію, Плюмбуму, Кадмію, Арсену) і їхні органічні сполуки. Вони блокують, наприклад, SH-групи (цистеїну), що входять у каталітичну ділянку ферменту. Неконкурентними інгібіторами є, наприклад, ціаніди, які міцно
93