Материал: Melnuchuk_Promuslova biotexnologij

РОЗДІЛ 5. ПРИНЦИПИ ТА МЕТОДИ ОТРИМАННЯ ПРОМИСЛОВИХ ШТАМІВ ПРОДУЦЕНТІВ
більше того, визначити різноманітність мікроорганізмів, існуючих в будьякій еконіші (екотопі) без попереднього культивування. Існує досить багато й інших методів, що дозволяють охарактеризувати мікробне різноманіття, і в кожному конкретному випадку вибір методу повинен визначатися поставленим завданням.
Як і в традиційних мікробіологічних методах, в молекулярнобіологічних методах є свої обмеження і умовності. Як неможливо виділити на одному середовищі і в одних умовах в чисті культури сильно відмінні за метаболізмом бактерії, так і, наприклад, для виявлення еубактерій і архей молекулярно-біологічними методами потрібні різні праймери. Для ізоляції НК рекомендується використовувати мінімальну кількість зразка, наприклад 0,5 г грунту, і, отже, виявлене розмаїття буде відображати екотопи розміром у 0,5 г.
При ідентифікації мікроорганізмів із природних зразків без виділення в чисті культури передбачається, що після проведення ПЛР отримують приблизно рівну кількість амплікон всіх присутніх у зразку бактерій. Однак це може бути далеко не так. У результаті проведення ПЛР з універсальним праймером отримують більше амплікон тих бактерій, чисельність яких була більше в початковому зразку і НК яких із зразка було виділено більше. При поділі продуктів ПЛР-методом ДГГЕ через дуже низьку концентрацію деяких амплікон їх смуги можуть бути важко помітні на тлі інших смуг. З іншого боку, високі концентрації амплікон близькоспоріднених організмів на недостатньо якісній ДГГЕ-фореграмі можуть виглядати у вигляді однієї об'ємної смуги. Крім цих проблем, що легше чи важче перевіряються та розв’язуються, але перевіряються й розв'язуваних, існують і більш важкі завдання, такі, як спонтанний синтез химерних амплікон при проведенні ПЛР, які легко можуть бути прийняті за наявність в зразку нових, неідентифікованих або некультивованих організмів. Зокрема, і через цей факт Міжнародним комітетом по систематиці і таксономії бактерій було прийнято рішення не приймати до розгляду опис нових мікроорганізмів без виділення чистих культур цих організмів.
У результаті проведення всіх стандартних операцій будуть отримані дані, що відображають тільки різноманітність мікроорганізмів у дослідженому зразку за типом «є-ні», але не кількісна присутність кожного виду. Формально вид, представлений у зразку однією клітиною і мільйоном клітин, має рівні шанси бути представленим у списку. Для подолання цієї істотної незручності в даний час розробляються методи кількісної ПЛР. В одному з них використовується внутрішній контроль у вигляді відомих концентрацій НК. Такі стандартні контрольні зразки підлягають ампліфікації поряд з експериментальними. Після проходження відповідної кількості циклів контрольні та експериментальні зразки порівнюються, а кількість НК і число циклів дають можливість
86

РОЗДІЛ 5. ПРИНЦИПИ ТА МЕТОДИ ОТРИМАННЯ ПРОМИСЛОВИХ ШТАМІВ ПРОДУЦЕНТІВ
розрахувати вихідну кількість того чи іншого мікроорганізму. Однак цей метод має ряд істотних недоліків через високу чутливість кінцевих етапів ПЛР до різного роду хімічних і фізичних факторів.
В іншому, більш точному методі, де використовується TaqMan 5'- нуклеази, має місце визначення накопичення ПЛР-продукту протягом реакції ампліфікації по зміні світіння флуоресцентної проби. При цьому відбувається накопичення флуоресцентного продукту, що призводить до посилення флуктуації-оресценціі, яка пропорційна накопиченню ПЛРпродукту. У цьому методі фактично контролюється кількість циклів, а не кількість ПЛР-продукту після фіксованої кількості циклів. Цей метод і його деякі модифікації отримали назву Real-Time PCR.
5.1.1. Руйнування клітин, виділення і очищення ДНК
Перший етап при ідентифікації мікроорганізмів молекулярнобіологічними методами - руйнування клітин і виділення НК. Відома велика кількість методів, що дозволяють руйнувати бактеріальні клітини як чистих культур, так і мікробних спільнот в зразках природних субстратів. В цілому слід зазначити, що для руйнування клітин чистих культур частіше використовують комбінацію детергентів і літичних ферментів, а для руйнування клітин в природних зразках (грунті, воді та ін.) - механічне руйнування. У тому випадку, якщо збираються проводити ідентифікацію бактерій, представлених колоніями різних розмірів на агаризованому середовищі, для зменшення наступних похибок рекомендується не просто змивати всі колонії, а відібрати рівну кількість біомаси кожній колонії. Відомо, що з 0,1 г сирої бактеріальної біомаси може бути виділено 40 - 200 мкг ДНК в залежності від виду бактерії і умов вирощування. При цьому зі збільшенням обсягу біомаси, використовуваної для виділення ДНК, ефективність виділення знижується.
При виділенні НК з природних зразків, об'єм зразка, використовуваного для виділення НК, залежить від передбачуваної великої кількості в ньому мікроорганізмів, а також типу зразка - вода, грунт або ін. Так як в природних зразках, особливо в грунті, часто присутні досить багато гумінових речовин, мінімізація зразка визначається і цим чинником. Зазвичай у разі грунтових зразків використовують 0,5 - 2,0 г грунту (у перерахунку на сухий стан зразка).
Послідовність операцій при руйнуванні клітин і екстрагуванні НК може бути наступною:
1. У пластикову пробірку на 15 мл вносять 2 г грунту, заливають 3 мл фосфатного буфера (120 мМ Na2HP04/NaH2P04 з рН 8,0), додають 3 г скляних стерильних бус розміром 0,1 мм. Закривають пробірку відповідної пробкою і поміщають її на 30 с (або два рази по 15 с) під струшувач (Bead Beater, Brown, Germany). У відсутність спеціального струшувачу суспензію
87

РОЗДІЛ 5. ПРИНЦИПИ ТА МЕТОДИ ОТРИМАННЯ ПРОМИСЛОВИХ ШТАМІВ ПРОДУЦЕНТІВ
зразку в буфері готують у стерильній порцеляновій ступці. Зразок заморожують рідким азотом і пестиком розтирають протягом приблизно 5 хв. до отримання гомогенної маси. Метод застосовують також і для руйнування грибних гіф.
2.Виділення тотальної фракції нуклеїнових кислот зазвичай здійснюють після додавання додецилсульфату натрію (від 30 мкл 10%-го розчину), ретельного перемішування та наступного внесення (від 0,5 мл) холодного водного (30%-го, рН 7,5 - 8,0) розчину фенолу (Duineveld et al., 2001). Суміш перемішують в руках (без використовування Вортекс) і витримують на льоду, після чого центрифугують на протязі 5 хв. на центрифузі при 15 тис. об/хв. при кімнатній температурі. При цьому вміст пробірки розшаровується на верхню водну фракцію і нижню - фенольну.
3.Піпеткою відбирають верхню частину супернатанту, намагаючись не захопити нижню фенольну частину. Відібрану верхню частину змішують з рівним об'ємом суміші хлороформу і ізоамілового спирту (24:1), перемішують в руках і відбирають водну фазу.
4.Додають до відібраної водної фази ОД обсягом 5 М NaCl і два об'єми холодного 96%-го етанолу. Суміш витримують на льоду протягом
20 хв. і 1 год. при -20°С.
5.Після цього суміш знову центрифугують 10 хв. на центрифузі при 15 тис. об/хв., але з охолодженням до -6°С.
6.Видаляють супернатант і промивають осад 1,5 мл 70%-го холодним етанолом. Осад виділеної ДНК повинен бути безбарвним (білим). Якщо ізольована ДНК залишається забарвленою (різної інтенсивності коричневий відтінок), то промивання спиртом повторюють.
Уцій же операції відбувається і видалення солі.
7.Відмитий зразок високополімерної ДНК підсушують на повітрі. Після цього суху ДНК розчиняють в деіонізованій воді і зберігають у замороженому вигляді при -20°С.
Частина отриманого препарату ДНК може бути використана для визначення процентного складу ГЦ-основ ДНК і ступеня ДНК-ДНК
гомології, якщо ДНК була виділена з чистої культури. Якщо тотальний препарат ДНК був виділений з зразків природних субстратів, особливо грунту, компостів та ін., фактично обов'язковою додатковою стадією є очищення ДНК. Для цієї мети краще використовувати набори реактивів і пристосування (колонки), що випускаються відповідними компаніями. Одна з них - Wizard DNA cleanup system (Promega, USA). Інструкція по застосуванню набору реактивів для додаткової очистки ДНК додається до кожного набору.
88

РОЗДІЛ 5. ПРИНЦИПИ ТА МЕТОДИ ОТРИМАННЯ ПРОМИСЛОВИХ ШТАМІВ ПРОДУЦЕНТІВ
5.1.2. Ампліфікація фрагментів виділеної та очищеної ДНК за допомогою ПЛР
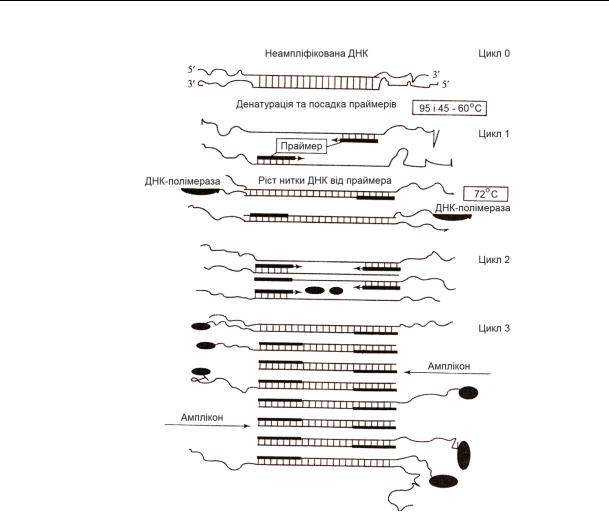
Для ампліфікації (збільшення числа копій) окремих фрагментів або сіквенсів (послідовностей) ДНК застосовують полімеразну ланцюгову реакцію (ПЛР). Вона відбувається, коли два олігонуклеотидних праймера гібридизуються на дволанцюговій матриці ДНК з двох різних кінців (5' і 3'), що дозволяє ДНК-полімеразі синтезувати ДНК-послідовність, що знаходиться між цими двома праймерами (рис.29).
Для проведення ПЛР необхідно мати: дволанцюгову ДНК-матрицю, що містить ділянку, яка повинна піддатися ампліфікації; праймери (в надлишку!), що представляють собою одноланцюгові фрагменти НК, які можуть «віджигати» (роз'єднувати) нитки ДНК, зв'язуючись з того або іншого кінця з одним з ланцюгів ДНК-матриці, комплементарної праймерам ділянки; суміш (надлишок!) чотирьох дезоксирибонуклеотидфосфатів (ДНФ), з яких буде синтезуватися нова ДНК; ДНК-полімеразу - фермент, який здійснює синтез нового фрагмента ДНК, амплікона. В ПЛР зазвичай використовують Taq-ДНК-полімеразу, високотермостабільний фермент, отриманий з термофільної бактерії Termus aquaticus. Ця полімераза запатентована фірмою, що її виробляє. Крім цього у реакційну суміш вносять також деякі необхідні компоненти для стабільності протікання реакцій.
ПЛР-продукти, що характеризують більшість грунтових бактерій, можна отримати з використанням бактеріальних праймерів U968 і L1401 і схеми сполуки-від'єднання праймерів протягом 30 теплових циклів. 17мірні праймери U968 і L1401 представляють собою нуклеотидні послідовності, специфічні висококонсервативні 16S РДНК області прокаріотів і відповідні положенню у Е. colі між 968 і 984; 1385 і 1401 нуклеотидами хромосомної ДНК відповідно.
Структура праймерів: U968 + GC: sequence 5 '- (GC clamp)-AAC GCG AAG AAC CTT АС-ЗЧ L140: sequence 5' - CGG TGT GTA CAA GAC CC-3 '.
Компонент GC clamp (зажим, доважок), являє собою 40-мірний фрагмент ДНК (5'-CGC CCG GGG CGC GCC CCG GGC GGG GCG GGG GCA CGG GGG G-3 '), призначений для розділення продуктів ПЛР методом ДГГЕ. Типова робоча суміш (master mix), використовувана для ампліфікації грунтових і ризосферних бактерій, містить: 23,34 мкл деіонізованої води; 5 мкл спеціального буфера (stoffel buffer), що містить 100 мМ трис-НС1, 100 мМ КС1, рН 8,3; 7,5 мкл (25мм) MgCl2; 10 мкл суміші чотирьох нуклеотидтрифосфатів: АТФ, ГТФ, ТГФ і ЦТФ; 1 мкл (10 мМ) U968 GC праймера; 1 мкл (10 мМ) L1401 праймера; 0,5 мкл 100%-го формамід; 0,16 мкл білка Т4 гена 32 (білок, що сприяє більш стабільному протіканню реакції); 0,5 мкл AmpliTaq DNA Polymerase, Stoffelfragment (10 од/мкл); 1
мкл ампліфікованої ДНК. Загальний обсяг суміші становить в даному
89
РОЗДІЛ 5. ПРИНЦИПИ ТА МЕТОДИ ОТРИМАННЯ ПРОМИСЛОВИХ ШТАМІВ ПРОДУЦЕНТІВ
Рис. 29. Схема полімеразної ланцюгової реакції (ПЛР)
(за О.С. Конічевим та Г.А. Севастьяновою, 2003)
випадку 50 мкл (van Dicpeningcn ct al., 2003). Готову суміш до внесення зразка ампліфікуємої ДНК піддають УФ-опроміненню протягом 3 хв. для стерилізації розчину.
Синхронність в роботі ДНК-полімерази і праймерів досягається за рахунок циклічної зміни температури суміші. Циклічну зміну температури забезпечує прилад ампліфікатор, що працює відповідно до введеної в нього програми. Після кожного циклу в реакцію включається і продукт (амплікон) попередньої реакції в якості ДНК-матриці. У результаті має місце геометрична прогресія збільшення кількості амплікон і після 30 - 40 циклів їх концентрація досягає мільйонів копій (амплікон). Точність відтворення амплікону забезпечується в істотному ступені якістю праймерів.
Надійним апліфікатором, рекомендованим для роботи, є прилад РТС200 (DNA Engine (Gradient cycler) BiOzym. The Netherlands) і відповідна програма. Послідовність циклів нагрівання-охолодження та їх тривалість може бути наступна. Початкову денатуруючу стадію проводять при температурі 94°С протягом 3 хв.; далі в кожному циклі денатурація
90