Материал: Chast1giper
– Нульовий рівень ентропії задається теоремою Нернста (німецький фізик і хімік), який у 1906 р встановив закон, який називають третім началом термодинаміки
![]() .
(6.69)
.
(6.69)
Ентропія системи при зменшенні її абсолютної температури до нуля теж зменшується до нуля.
– З’ясуємо
фізичний зміст ентропії. З першого
начала термодинаміки
![]() .
Нехай маємо ізотермічний оборотний
процес. Тоді
.
Нехай маємо ізотермічний оборотний
процес. Тоді
![]() ,
,
![]() .
(6.70)
.
(6.70)
Тут
![]() називається вільною енергією – це та
частина внутрішньої енергії, за рахунок
зменшення якої (знак мінус в 6.70) система
може виконати роботу. Тоді добуток TS
дає ту
частину внутрішньої енергії, яка не
може перетворитись в роботу, тобто
зв’язану енергію. Дійсно, якби в роботу
можна було перетворити всю внутрішню
енергію, це означало б зникнення системи.
називається вільною енергією – це та
частина внутрішньої енергії, за рахунок
зменшення якої (знак мінус в 6.70) система
може виконати роботу. Тоді добуток TS
дає ту
частину внутрішньої енергії, яка не
може перетворитись в роботу, тобто
зв’язану енергію. Дійсно, якби в роботу
можна було перетворити всю внутрішню
енергію, це означало б зникнення системи.
– У 1872 р. австрійський фізик-теоретик Л.Больцман (1844-1906) зв’язав ентропію з термодинамічною ймовірністю W стану системи
![]() .
(6.71)
.
(6.71)
Т![]() ут
k
– стала Больцмана, W
- термодинамічна
ймовірність
– це кількість мікроскопічних станів
систем, якими може бути реалізований
макроскопічний її стан. Н
ут
k
– стала Больцмана, W
- термодинамічна
ймовірність
– це кількість мікроскопічних станів
систем, якими може бути реалізований
макроскопічний її стан. Н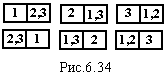 априклад,
макроскопічний стан системи із трьох
молекул газу – одна молекула знаходиться
в одній половині посудини, а дві в другій
може бути реалізований трьома способами
(рис.6.34). Стани, зображені один під одним
однакові, так як не уточнюється в якій
саме (лівій чи правій) знаходиться одна
молекула. Отже W
= 3.
априклад,
макроскопічний стан системи із трьох
молекул газу – одна молекула знаходиться
в одній половині посудини, а дві в другій
може бути реалізований трьома способами
(рис.6.34). Стани, зображені один під одним
однакові, так як не уточнюється в якій
саме (лівій чи правій) знаходиться одна
молекула. Отже W
= 3.
-
Зміна ентропії ідеального газу. Ізоентропійний (адіабатний) процес
Враховуючи співвідношення (6.67), (6.38), (6.41), (6.42) і (6.5), одержуємо зміну ентропії ідеального газу
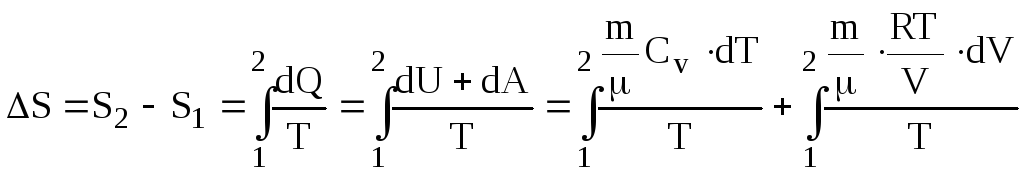
![]() .
(6.72)
.
(6.72)
Знайдемо зміну ентропії при адіабатному процесі. Для цього в (6.72) відношення температур заміняємо із рівняння адіабати (6.55)
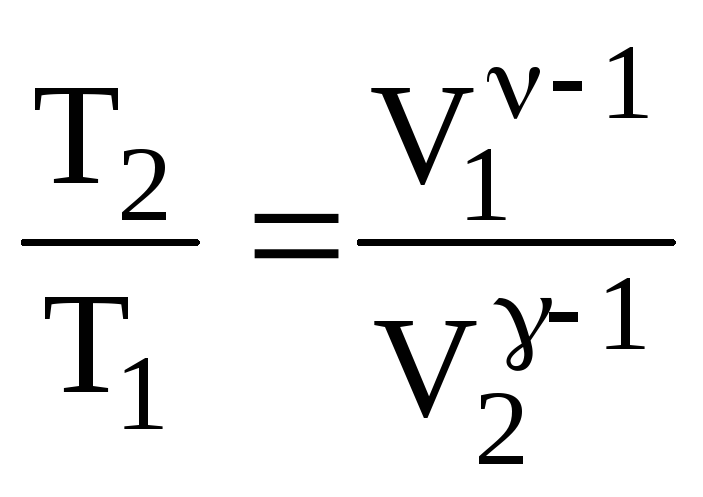 .
Одержуємо
.
Одержуємо
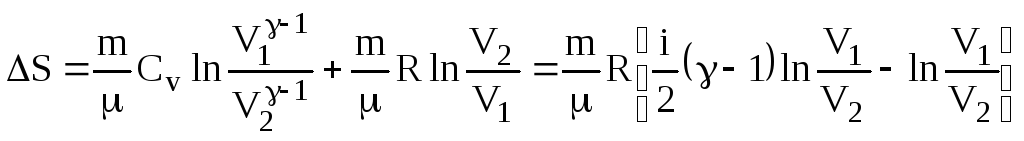 .
(6.73)
.
(6.73)
![]() .
Тому у (6.73) вираз у квадратній дужці
дорівнює нулю.
.
Тому у (6.73) вираз у квадратній дужці
дорівнює нулю.
Отже,
![]() .
А це означає, що ентропія при адіабатному
процесі не змінюється (залишається
сталою). Тому адіабатний процес називається
ще ізоентропійним.
.
А це означає, що ентропія при адіабатному
процесі не змінюється (залишається
сталою). Тому адіабатний процес називається
ще ізоентропійним.
-
Реальні гази. Рівняння Ван-дер-Ваальса та його аналіз. Зрідження газів
При низькій температурі і високому тискові має спостерігається відхилення газових процесів від законів ідеального газу. Газ стає реальним. Тепер необхідно враховувати: 1) власний об’єм молекул, наприклад, при тискові 100 ат вони займають (1÷2)%; 2) сили взаємодії між молекулами, головним чином сили притягування. Сили відштовхування проявляються на значно менших відстанях, що має місце в твердих тілах. За рахунок цих сил реальний газ уже стиснутий внутрішніми силами.
Враховуючи ці фактори, нідерландський фізик Ван-дер-Ваальс у 1873 р. вивів рівняння стану реального газу. Для 1 моля воно має вид
![]() .
(6.74)
.
(6.74)
Т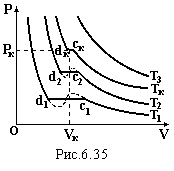
![]() ут:
ут:![]() - константи, Рі
– внутрішній тиск, Vм
– об’єм 1 моля газу, n – концентрація,
vo
– об’єм однієї молекули. Поправка b
враховує
власний об’єм молекул, за рахунок чого
об’єм, доступний для молекул фактично
менший, ніж об’єм посудини. Тому ця
поправка віднімається із об’єму. Щоб
записати рівняння для довільної кількості
газу, необхідно в (6.74) замінити
- константи, Рі
– внутрішній тиск, Vм
– об’єм 1 моля газу, n – концентрація,
vo
– об’єм однієї молекули. Поправка b
враховує
власний об’єм молекул, за рахунок чого
об’єм, доступний для молекул фактично
менший, ніж об’єм посудини. Тому ця
поправка віднімається із об’єму. Щоб
записати рівняння для довільної кількості
газу, необхідно в (6.74) замінити
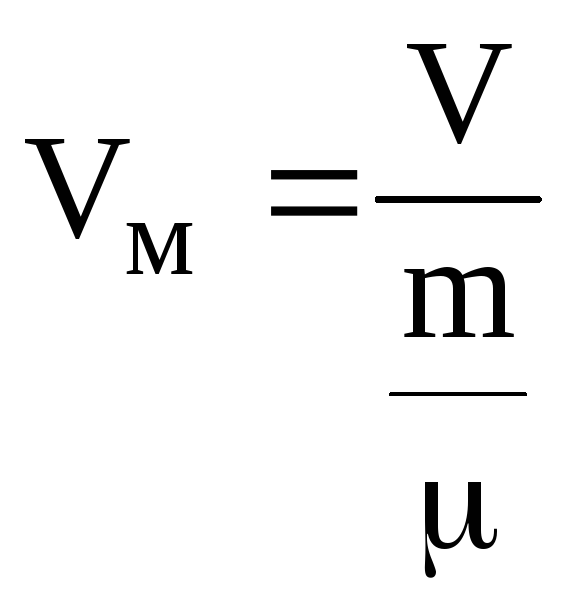 .
Після алгебраїчних спрощень одержуємо
рівняння Ван-дер-Ваальса для довільної
кількості газу
.
Після алгебраїчних спрощень одержуємо
рівняння Ван-дер-Ваальса для довільної
кількості газу
![]() .(6.75)
.(6.75)
Експериментальні
ізотерми реального газу зображені на
рис.6.35. Рівняння (6.75) добре описує ці
криві, за винятком горизонтального
участку конденсації (пунктирна лінія).
При зменшенні об’єму реального газу
його тиск зростає. В певному стані (точки
с) починається конденсація. Виникає
рідина і тиск дорівнює тискові насиченої
пари. При незмінній температурі він
залишається сталим, поки вся пара не
перейде в рідку фазу (точки d).
З ростом температури
ділянка cd
конденсації скорочується і при критичній
температурі ТК
вироджується в точку. Стан, який їй
відповідає, називається критичним,
коли в рівновазі знаходяться всі три
фази: тверда, рідка і газоподібна.
Параметри цього стану TК,
PК,
VК
для різних речовин різні. Наприклад,
для води: VК
= 3 см3,
PК
= 218 ат, TК
= 374оС.
При Т >
TК
рідка фаза не виникає. Отже, для зрідження
газу шляхом його стискання необхідно
знизити температуру нижче за критичну.
Після закінчення конденс![]() ації
стискується рідина, тому тиск різко
зростає.
ації
стискується рідина, тому тиск різко
зростає.
Л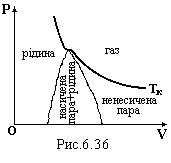
![]() інія,
яка з’єднує точки c
початку і точки d
кінця конденсації (рис.6.36) разом з
ізотермою при критичній температурі
ТК
поділяє Р-V
– діаграму на чотири області: 1) газ; 2)
ненасичена пара; 3) насичена пара + рідина;
4) рідина.
інія,
яка з’єднує точки c
початку і точки d
кінця конденсації (рис.6.36) разом з
ізотермою при критичній температурі
ТК
поділяє Р-V
– діаграму на чотири області: 1) газ; 2)
ненасичена пара; 3) насичена пара + рідина;
4) рідина.
-
Внутрішня енергія реального газу
Внутрішня енергія реального газу складається із кінетичної енергії теплового руху молекул і потенціальної енергії їх взаємодії між собою. Перша складова – це не що інше, як внутрішня енергія ідеального газу (6.41). Другу складову знайдемо як роботу газу проти внутрішнього тиску Рі. Для 1 моля газу
![]() .
.
Константу
інтегрування знайдемо із умови: при Vм
→ ∞ Uім
= 0 (Газ стає ідеальним). Одержуємо С = 0.
Отже внутрішня енергія 1 моля реального
газу дорівнює
![]() .
(6.76)
.
(6.76)
Знайдемо внутрішню енергію для декількох молей аналогічно (6.75)
![]() .
(6.77)
.
(6.77)
Одержаний вираз показує, що внутрішня енергія реального газу, на відміну від ідеального, являється функцією не тільки температури, а і об’єму. При розрідженні реальний газ переходить у ідеальний, другий доданок перетворюється в нуль, і формула (6.77) переходить в (6.41), як і повинно бути.
-
Рідини. Явища в рідинах
П
![]() о
характеру руху молекул і сил взаємодії
між ними рідини займають проміжне
положення між газами і твердими тілами.
В рідинах молекули протягом певного
часу здійснюють коливання навколо
тимчасового положення рівноваги. Цей
час називається часом осідлого стану
молекули. Потім молекула перескакує в
інше положення рівноваги (рис.6.37). Ці
хаотичні переходи нагадують рух молекул
газу, а коливальний рух – рух атомів у
твердих тілах. Для рідин характерний
ближній порядок в розміщенні молекул.
Це означає, що розміщення найближчих
сусідніх молекул однакове для всіх
молекул. Але по мірі віддалення такий
порядок порушується. Твердим же
кристалічним тілам характерний дальній
порядок в розміщенні молекул. У зв’язку
з цим для кристалів має місце анізотропія
властивостей (різні властивості в різних
напрямках), а для рідин характерні
ізотропні
(однакові в різних напрямках) властивості.
Але існують так звані рідкі кристали,
названі так із-за анізотропії своїх
властивостей, яка зумовлена анізотропією
властивостей окремих молекул, а не
дальнім порядком в їх розміщенні.
Молекули рідких кристалів уявляють
собою довгі ланцюги полімерних сполук.
При паралельній одна одній орієнтації
молекул і виникає анізотропія.
о
характеру руху молекул і сил взаємодії
між ними рідини займають проміжне
положення між газами і твердими тілами.
В рідинах молекули протягом певного
часу здійснюють коливання навколо
тимчасового положення рівноваги. Цей
час називається часом осідлого стану
молекули. Потім молекула перескакує в
інше положення рівноваги (рис.6.37). Ці
хаотичні переходи нагадують рух молекул
газу, а коливальний рух – рух атомів у
твердих тілах. Для рідин характерний
ближній порядок в розміщенні молекул.
Це означає, що розміщення найближчих
сусідніх молекул однакове для всіх
молекул. Але по мірі віддалення такий
порядок порушується. Твердим же
кристалічним тілам характерний дальній
порядок в розміщенні молекул. У зв’язку
з цим для кристалів має місце анізотропія
властивостей (різні властивості в різних
напрямках), а для рідин характерні
ізотропні
(однакові в різних напрямках) властивості.
Але існують так звані рідкі кристали,
названі так із-за анізотропії своїх
властивостей, яка зумовлена анізотропією
властивостей окремих молекул, а не
дальнім порядком в їх розміщенні.
Молекули рідких кристалів уявляють
собою довгі ланцюги полімерних сполук.
При паралельній одна одній орієнтації
молекул і виникає анізотропія.
В рідинах спостерігаються ряд специфічних для них явищ:
1) поверхнева енергія; 2) сила поверхневого натягу; 3) поверхневий тиск; 4) змочування і незмочування 5) капілярні.
Розглянемо кожне із цих явищ.
–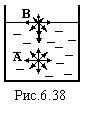
![]() Поверхнева
енергія.
Розглянемо
сили, які діють з боку сусідніх молекул
на дві молекули рідини (рис.6.38): об’ємну
(А) і поверхневу (В). Оточення об’ємної
молекули А симетричне, тому рівнодіюча
сил дорівнює нулю. На поверхневу молекулу
В діють сили з боку рідини більші, ніж
з боку газу. Виникає рівнодіюча сила,
направлена всередину рідини. Отже для
переведення молекули з об’єму на
поверхню необхідно виконати роботу
проти цієї рівнодіючої. Ця робота
перетворюється в потенціальну енергію
поверхневих молекул.
Поверхнева
енергія.
Розглянемо
сили, які діють з боку сусідніх молекул
на дві молекули рідини (рис.6.38): об’ємну
(А) і поверхневу (В). Оточення об’ємної
молекули А симетричне, тому рівнодіюча
сил дорівнює нулю. На поверхневу молекулу
В діють сили з боку рідини більші, ніж
з боку газу. Виникає рівнодіюча сила,
направлена всередину рідини. Отже для
переведення молекули з об’єму на
поверхню необхідно виконати роботу
проти цієї рівнодіючої. Ця робота
перетворюється в потенціальну енергію
поверхневих молекул.
Поверхнева
енергія
US
дорівнює різниці енергії поверхневих
молекул і енергії такої ж кількості
об’ємних молекул. Ясно, що вона пропорційна
кількості поверхневих молекул, тобто
площі поверхні рідини S
![]() .
(6.78)
.
(6.78)
Тут
![]() - коефіцієнт
поверхневого натягу, для кожної рідини
величина стала, але залежить від
температури і домішок.
- коефіцієнт
поверхневого натягу, для кожної рідини
величина стала, але залежить від
температури і домішок.
–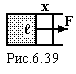 Сила
поверхневого натягу.
Відомо
(розділ 4.7), що стійкою рівновагою системі
є стан з мінімальною потенціальною
енергією. Тому рідина має тенденцію
зайняти стан з мінімальною площею
поверхні, тобто скоротитись. Це приводить
до виникнення сили поверхневого натягу,
яка діє вздовж межі поверхні по дотичній
до неї. Знайдемо величину цієї сили F.
Нехай н
Сила
поверхневого натягу.
Відомо
(розділ 4.7), що стійкою рівновагою системі
є стан з мінімальною потенціальною
енергією. Тому рідина має тенденцію
зайняти стан з мінімальною площею
поверхні, тобто скоротитись. Це приводить
до виникнення сили поверхневого натягу,
яка діє вздовж межі поверхні по дотичній
до неї. Знайдемо величину цієї сили F.
Нехай н![]() а
прямокутну рамку з рухомою стороною
а
прямокутну рамку з рухомою стороною
![]() натягнута плівка рідини (рис.6.39).
Розтягнемо плівку силою F
на відстань х. Таке розтягування фактично
є не що інше, як процес переводу молекул
із об’єму рідини на поверхню. Буде
виконана робота А = F∙x,
яка дорівнює з
натягнута плівка рідини (рис.6.39).
Розтягнемо плівку силою F
на відстань х. Таке розтягування фактично
є не що інше, як процес переводу молекул
із об’єму рідини на поверхню. Буде
виконана робота А = F∙x,
яка дорівнює з![]() більшенню
поверхневої енергії
більшенню
поверхневої енергії
![]() .
Одержуємо для сили поверхневого натягу
.
Одержуємо для сили поверхневого натягу
![]() .
(6.79)
.
(6.79)
Силою поверхневого натягу рідина уже стиснута. Цим і пояснюється погана стискуваність рідин.
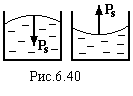
–
![]() Поверхневий
тиск.
Коли поверхня рідини викривлена
(утворився меніск), її площа більша, ніж
плоскої поверхні і поверхнева енергія
не мінімальна. Тенденція поверхні до
скорочення приводить до виникнення
сили, яка направлена до центру кривизни
поверхні. Виникає поверхневий тиск РS
(рис.6.40). Знайдемо його величину на
прикладі краплини рідини, яка має форму
кулі радіусом R
(таку форму
буде мати рідина у стані невагомості).
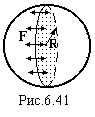
Уявно розріжемо її діаметральною
площиною (рис.6.41). Вздовж лінії перерізу
(кола) діє сила поверхневого натягу
Поверхневий
тиск.
Коли поверхня рідини викривлена
(утворився меніск), її площа більша, ніж
плоскої поверхні і поверхнева енергія
не мінімальна. Тенденція поверхні до
скорочення приводить до виникнення
сили, яка направлена до центру кривизни
поверхні. Виникає поверхневий тиск РS
(рис.6.40). Знайдемо його величину на
прикладі краплини рідини, яка має форму
кулі радіусом R
(таку форму
буде мати рідина у стані невагомості).
Уявно розріжемо її діаметральною
площиною (рис.6.41). Вздовж лінії перерізу
(кола) діє сила поверхневого натягу
![]() ,
яка і стискує дві півкулі. Ця сила
розподіляється по площі перерізу
,
яка і стискує дві півкулі. Ця сила
розподіляється по площі перерізу
![]() .
Виникає поверхневий тиск, направлений
до центра кулі
.
Виникає поверхневий тиск, направлений
до центра кулі
![]() .
(6.80)
.
(6.80)
Т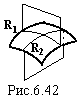
![]() ут
ут
![]() - кривизна сферичної поверхні. Якщо ж
поверхня не сферична, то її кривизна
визначається як півсума обернених
радіусів кривизни R1
і R2
ліній перетину цієї поверхні двома
будь-якими взаємно-перпендикулярними
площинами (рис.6.42)
- кривизна сферичної поверхні. Якщо ж
поверхня не сферична, то її кривизна
визначається як півсума обернених
радіусів кривизни R1
і R2
ліній перетину цієї поверхні двома
будь-якими взаємно-перпендикулярними
площинами (рис.6.42)
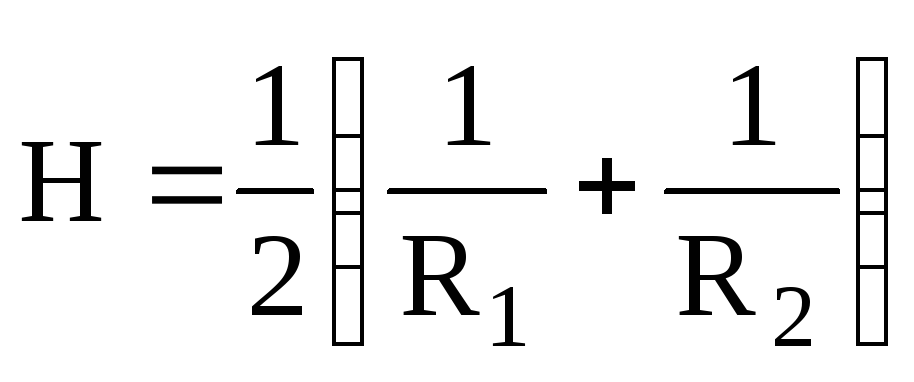 .
(6.81)
.
(6.81)
Н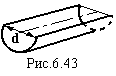
![]() априклад,
для циліндричного меніска (рис.6.43)
кривизна
априклад,
для циліндричного меніска (рис.6.43)
кривизна
![]() .
.
–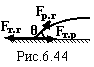
![]() Явище
змочування і не змочування.
На поверхневу
молекулу, яка межує з поверхнею твердого
тіла і газом діють сили: Fт.р
– на межі тверда поверхня-рідина; Fт..г
– на межі тверде тіло-газ; Fр..г
– на межі рідина-газ (рис.6.44). Умовою
рівноваги цієї молекули є рівняння
Явище
змочування і не змочування.
На поверхневу
молекулу, яка межує з поверхнею твердого
тіла і газом діють сили: Fт.р
– на межі тверда поверхня-рідина; Fт..г
– на межі тверде тіло-газ; Fр..г
– на межі рідина-газ (рис.6.44). Умовою
рівноваги цієї молекули є рівняння