Материал: Сыровая А.О. и др Аминокислоты глазами химиков, фармацевтов, биологов. Т. 2
Фенилаланин
Синтез белка |
Фенилаланиноксидаза |
Фенилпировиноградная Фенилуксусная Фенилмолочная
Мозг |
Печень |
Почки |
(накопление) |
(накопление) |
(выделение) |
|
|
Фенилгеконовые тела |
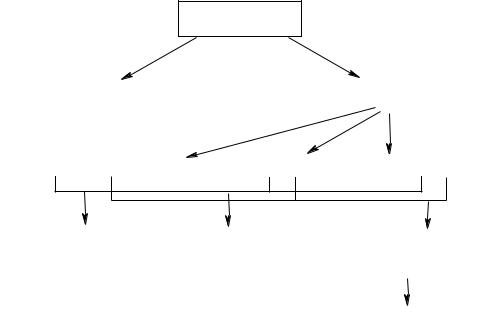
Рис.6. Катаболизм фенилаланина в организме В результате нарушается миелинизация нервных волокон, что ведет к
нарушению развития нервной системы и слабоумию – олигофрении вплоть до идиотии. Наиболее драматичным является то, что фенилаланин младенец получает с молоком матери. Если диагностика была проведена своевременно,
вред фенилаланина можно нейтрализовать, значительно ограничив (у педиатров есть нормативные таблицы) его поступление в организм на этапе от рождения до полового созревания. В этом случае необратимых патологических изменений тканей мозга можно полностью избежать: ребенок вырастет полноценным.
Следствием нарушенного обмена в мозге является тяжелое психическое недоразвитие.
Поступающий в организм фенилаланин идет на построение белковой цепи или превращается в тирозин. Отсутствие в печени фермента фенилаланингидроксидазы препятствует нормальному превращению фенилаланина пищи в тирозин.
Поэтому фенилаланин используется лишь при синтезе белка, а избыток накапливается в клетках печени и попадает в кровоток, где количество фенилаланина является токсичным для клеток мозга. Почки не справляются с его
26
реабсорбцией, в результате чего он выводится с мочой. Именно наличие этого фенилкетона в моче дало основание назвать соответствующее патологическое состояние фенилкетонурией [22]. Как ни прискорбно, фенилаланин вреден при фенилкетонурии. В печени здоровых людей небольшая часть фенилаланина
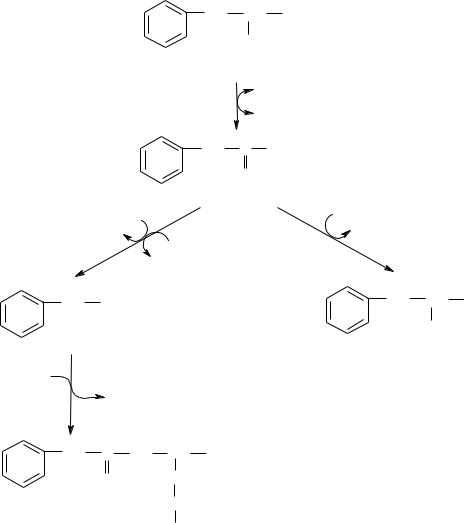
( 10%) превращается в фениллактат и фенилацетилглутамин (см. рис.7). Этот путь катаболизма фенилаланина становится главным при нарушении основного пути – превращения в тирозин, который катализируется фенилаланин-4-
гидроксилазой. При дефиците этого фермента накопившийся фенилаланин подвергается трансаминированию с α-кетоглутаратом. Нарушение проявляется в раннем младенческом возрасте и приводит к накоплению фенилаланина, т.к.
тирозин не синтезируется организмом младенца, и это еще больше замедляет удаление избытка фенилаланина, который превращается в фенилацетат,
фенилацетилглутамин, фенилпируват, фениллактат, орто-гидроксифенилацетат
(рис.7). Эти соединения токсичны для клеток мозга.
Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути.
Этиология и патогенез. Гиперфенилаланинемии – это группа генетически гетерогенных аутосомно-рецессивных заболеваний, обусловленных нарушением метаболизма фенилаланина. В основе патогенеза гиперфенилаланинемии лежит накопление в крови фенилаланина и продуктов его утилизации:
фенилпировиноградной, фенилмолочной и фенилуксусной кислот, оказывающих токсический эффект на различные органы и ткани, в первую очередь на головной мозг (рис. 7).
Фенилкетонурия, наиболее частая и злокачественная форма гиперфенилаланинемии.
27
|
|
CH2 |
CH |
COOH |
|
|
|
NH2 |
|
|
|
L-Фенилаланин |
||
|
|
|
a-КГ |
|
|
|
ПФ |
|
|
|
|
|
Глу |
|
|
|
CH2 |
C |
COOH |
|
|
|
O |
|
|
|
Фенилпируват |
||
|
NAD+ |
|
NADH+H+ |
|
|
|
|
||
NADH+H+ |
|
|
NAD+ |
|
|
|
Н2 О |
|
|
|
|
СО2 |
|
|
CH |
COOH |
|
|
CH2 CH COOH |
2 |
|
|
|
|
Фенилацетат |
|
|
ОН |
|
|
|
Фениллактат |
||
|
|
|
|
|
Глн |
|
|
|
|
|
Н2 О |
|
|
|
CH2 |
C NH |
CH COOH |
|
|
|
O |
CH2 |
|
|
CH2
Фенилацетилглутамин CONH2
Рис.7. Альтернативные пути катаболизма фенилаланина.
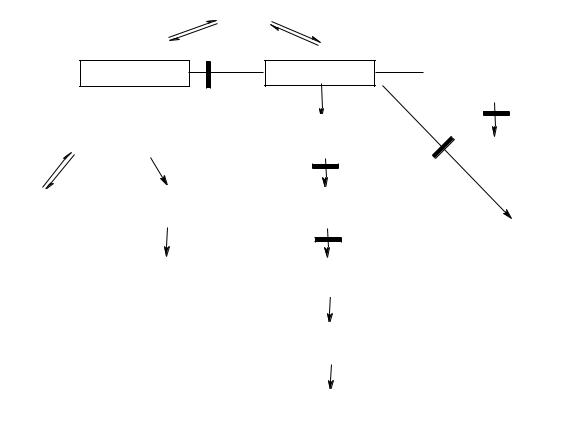
Нарушения обмена фенилаланина. Фенилаланин в норме необратимо окисляется в тирозин. Если же в печени нарушается синтез необходимого для этого фермента фенилаланингидроксилазы (см. рис. 8, блок а), то окисление фенилаланина идет по пути образования фенилпировиноградной и фенилмолочной кислот — развивается фенилкетонурия. Однако этот путь обладает малой пропускной способностью и поэтому фенилаланин накапливается в большом количестве в крови, тканях и цереброспинальной жидкости, что в первые же месяцы жизни ведет к тяжелому поражению центральной нервной системы и неизлечимому слабоумию.
28
Из-за недостаточного синтеза тирозина снижается образование меланина,
что обусловливает посветление кожи и волос. Кроме того, при увеличенной выработке фенилпировиноградной кислоты тормозится активность фермента
(дофамингидроксилазы), необходимого для образования катехоламинов
(адреналина, норадреналина). Поэтому тяжесть наследственного заболевания определяется комплексом всех этих нарушений.
Установить болезнь можно с помощью следующей пробы: при добавлении к свежей моче нескольких капель 5% раствора трихлоруксусного железа появляется оливково-зеленая окраска. Больные погибают в детстве, если не проводится специальное лечение, которое заключается в постоянном, но осторожном (контроль за аминокислотным составом крови) ограничении поступления фенилаланина с пищей (см. рис.8).
|
Белки пищи и тканей |
|
||
|
а |
|
Диоксифенилаланин |
|
Фенилаланин |
Тирозин |
|||
(ДОФА) |
||||
|
|
|
||
Фенилпировино- |
n-Гидроксифенилпиро- |
д |
||
г |
||||
граднаякислота |
винограднаякислота |
Меланин |
||
|
|
|
||
|
|
б |
|
|
Фениллактат |
Фенилуксусная |
Гомогентизиновая |
|
|
|
кислота |
кислота |
Тироксин |
|
|
|
в |
||
|
|
трийодтиронин |
||
|
|
|
||
|
Фенилацетил- |
Малеилацетоуксусная |
|
|
|
глутамин |
кислота |
|
|
|
|
Фумарилацетоуксусная |
|
|
|
|
кислота |
|
|
Фумаровая+ ацетоуксуснаякислота
Рис. 8. Блокада путей метаболизма фенилаланина и тирозина
29
На рис.8 изображены биохимические превращения фенилаланина и тирозина и основные метаболические блоки на их пути.
Ведущим симптомом болезни является отставание умственного развития,
достигающее у большей части больных степени имбецильности или идиотии. Уже с первых недель жизни ребенка наблюдаются повышенная возбудимость,
повышение рефлексов, мышечная ригидность и судорожный синдром. Одним из первых неспецифических проявлений заболевания может быть повторяющаяся рвота. В 80-90% наблюдений у детей выражен дефект пигментации,
обусловленный дефицитом меланина. Большинство из них блондины с голубыми глазами и светлой кожей. Нередки мокнущие экземы и дерматиты. Всем новорожденным проводится обязательное централизованное специальное скринирующее исследование для выявления среди них больных фенилкетонурией. Иллюстрацией результативности Налаженный скрининг совсем не исключает проведение диагностических мероприятий по выявлению фенилкетонурии среди групп детей повышенного риска. Таковыми являются умственно неполноценные дети, находящиеся в специализированных учреждениях; дети, отстающие в умственном развитии, с поражениями кожных покровов; а также братья и сестры больных фенилкетонурией.
В результате мутации гена, контролирующего синтез фенилаланингидроксилазы, развивается метаболический блок на этапе превращения фенилаланина в тирозин, вследствие чего основным путем преобразования фенилаланина становится дезаминирование и синтез токсических производных – фенилпировиноградной, фенилмолочной и фенилуксусной кислот
(рис. 8). В крови и тканях значительно увеличивается содержание фенилаланина
(до 0,2 г/л и более при норме 0,01-0,02 г/л). Существенную роль в патогенезе болезни играет недостаточный синтез тирозина, который является предшественником катехоламинов и меланина. Заболевание наследуется по аутосомно-рецессивному типу.
30