Материал: Молекулярно-генетическая характеристика рекомбинантных форм вируса иммунодефицита человека 1 типа, выявленных на территории Республики Беларусь
Годом позже, в 2008-м, Ч. Рива с соавторами выдвинул гипотезу о том, что AFSUпроизошел из Гвинеи. Они выявили ВИЧ-1 А1 (под именем 60 000), который был генетически очень близок кAFSU. Вирус был найден у пациента из г. Конакри, столицы Республики Гвинея, который иммигрировал в Италию. Данный изолят также находился в основании кластера, сформированного вариантами AFSU[162].
В 2015 г. Франциско Диез-Ферте с соавторами[163] повторили исследования о происхождении AFSU с использованием байесовских филодинамических подходов, основанных на теории коалесценции, и вывели наиболее вероятную эволюционную гипотезу о происхождении AIDU. Согласно их исследованиям, AFSUимеет достоверное происхождение из Демократической Республики Конго, постериорная достоверность байесовской статистки составила 0,84 по анализу фрагмента C2-V2-V3. Гипотеза, предложенная Ч. Ривой в 2008 г., не нашла подтверждения. Так, постериорная достоверность кластера с изолятом 60 000 из Республики Гвинея составила всего 0,0003.
Филогеографический анализ фрагмента p24gag выявил, что AFSU-вариант появился в Одессе приблизительно в 1984 г. (1982-1987гг.), то есть приблизительно за 10 лет до начала распространения среди инъекционных наркоманов. Изначально распространение вируса происходило преимущественно половым путем, о чем свидетельствуют образцы, расположенные у основания кластера и принадлежащие пациентам, которые были инфицированы гетеросексуально не позже 1993 г. Анализ фрагмента C2-V2-V3 гена env также указал на появление AFSUв г. Одессе в 1984 г. Комбинированные результаты обоих анализов указывают интервал для tMRCA (время наиболее близкого общего предка) AFSU- 1982-1987 гг.
Таким образом, вариант AFSUпоявился в украинском городе Одессе приблизительно в 1984 г., за 10 лет до начала эпидемии на территории стран бывшего СССР.
.10 Эпидемия ВИЧ-1 на территории Беларуси
Первый случай ВИЧ-инфекции на территории Беларусибыл выявлен в РНПЦ эпидемиологии и микробиологии в 1986 г. у студента из Бурунди. В 1990 г. был зарегистрирован первый случай СПИДа в стране у женщины - жительницы Беларуси [164]. С 1986 по 1996 гг. количество случаев ВИЧ/СПИД, выявляемых ежегодно в Беларуси, варьировало от 12 до 14, и на 1 января 1996 г. было официально зарегистрировано 113 случаев ВИЧ-инфекции. 66% всех случаев ассоциировано с половым путем передачи вируса.Молекулярно-эпидемиологические исследования показали доминирование генетически гетерогенного ВИЧ-1, главным образом, субтипа С [140]. Летом 1996 г. в г. Светлогорске Гомельской области был зафиксирован резкий рост случаев выявления ВИЧ-инфекции у лиц, являвшихся потребителями инъекционных наркотиков. Так, в июле 1996-го было зафиксировано 60 случаев ВИЧ-инфекции, а на 1 ноября 1997 г. общее число случаев ВИЧ-инфекции составило уже 1728, 85% из которых выявлено у ПИН. В отличие от Украины и России, где эпидемия ВИЧ-1 поразила различные географические регионы, в Беларуси эпидемия была ограничена в основном г. Светлогорском, на долю которого приходилось более 70%всех случаев ВИЧ-инфекции среди ПИН. В Светлогорске с населением в 72 000 человек ВИЧ-1 был диагностирован у более чем 1200 из 14 000 инъекционных наркоманов, из которых 88% являлись лицами в возрасте 18-29 лет. Молекулярные исследования показали, что вспышка была вызвана ВИЧ-1 субтипа А1, который генетически идентичен вирусам у ПИН в Южной Украине и Центральной и Южной России [140].
По состоянию на 1 декабря 2015г. в Республике Беларусь зарегистрировано 19 605 случаев ВИЧ-инфекции, количество людей, живущих с ВИЧ-1 - 15 069, показатель распространенности составил 158,9 на 100 тысяч населения[165].
За период 1996-2008 гг. на территории Беларуси по-прежнему доминирующим оставался ВИЧ-1 субтипа А1, вариант AFSU, на долю которого приходилось около 95% от всех случаев ВИЧ-инфекции. В четырех случаяхбыло выявлено инфицирование ВИЧ-1 субтипом В. Данные изоляты при филогенетическом анализе не были близкими между собой и, вероятно, явились результатом четырех эпидемиологически независимыхзаносов[166]. Помимо субтипов А1 и B, выявлены варианты ВИЧ-1 субтипов С, G, CRF02_AG, CRF03_AB, CRF06_cpx, а также уникальная рекомбинантная форма ВИЧ-1 AFSU/BFSU.
Филогенетическая систематика
Филогенетическая систематика, или филогенетика - наука, изучающая эволюционные отношения между разными видами на Земле. Эволюционный процесс можно рассматривать как процесс ветвления, и результаты обычно представлены в форме бифуркационного филогенетического дерева (phylogenetic tree), на котором ветви от двух любых таксонов соединены в одном узле. Порядок узлов и ветвление дерева составляют топологию филогенетического дерева, длины ветвей дерева соответствуют генетическому расстоянию между таксонами. Высокая скорость эволюционной изменчивости ВИЧделает его идеальной моделью для изучения эволюционного процесса и позволяет проследить эволюцию в режиме «реального времени» [167].
Эволюционный процесс включает в себя изменения генома в виде инсерций, делеций, замен нуклеотидов и рекомбинационные события. Эволюционные отношения изучаются методом попарного сравнения, поэтому первым и важным этапом филогенетического анализа является создание алаймента сиквенсов (sequence alignment) нуклеотидов или аминокислот. Далее выводы о филогенетических отношениях будут зависеть от выбранной модели нуклеотидных замен (substitution model) для данного алаймента, модели реконструкции дерева и, в конечном счете, от оценки статистической достоверности отношений на филогенетическом дереве.
Модели нуклеотидных замен
Два сиквенса (таксона), у которых один общий предок (узел), могут развиваться независимо друг от друга. Наиболее простой подход к измерению дивергенции между сиквенсами - расчет числа различий в сайте, обозначаемых генетической дистанцией, или p-дистанцией (p-distance). К сожалению, когда дивергенция очень высокая, p-дистанция не точно оценивает количество замен, произошедших на самом деле. Например, параллельная замена, при которой оба связанных таксона приобрели одну и ту же замену в одном и том же сайте, таким образом скрывая эволюционное событие, на самом деле произошедшее. Подобным образом влияют на недостоверный расчетp-дистанции и обратные мутации, например, А → С →А, в случае которых замена, на самом деле произошедшая, не учитывается. И, наконец, в сайте может произойти несколько замен: A → G → T, но только одна замена, A → T, будет учитываться. С целью более точного вычисления эволюционных изменений было предложено несколько моделей нуклеотидных замен. Все эти модели относятся к классу цепей Маркова с дискретным и непрерывным временем, в которых используется матрица интенсивностей Q для указания относительной скорости замены каждого нуклеотида на протяжении всей длинысиквенса.
Более того, все модели принимают во внимание следующие положения:
. скорость замены основания i на основание j не зависит от основания, которое находилось в данном сайте до i;
. скорость замен неизменна во времени;
. относительная скорость изменений нуклеотидов A,G,T,C - одинаковая.
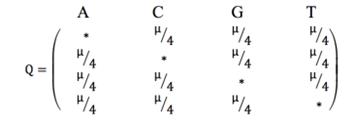
Наиболее простая модель нуклеотидных замен - модельJC69(Jukes and Cantor, 1969), которая предполагает, что все нуклеотиды имеют одинаковую частоту появления (25% каждый): πA= πG= πT= πC= ¼, где π - частота появления каждого нуклеотида, и каждый нуклеотид имеет равную вероятность быть замененным любым другим (рисунок 1.8)[168].
В модели JC69 частота замены всех нуклеотидов равна 1:a=b=c=d=e=
= f=1. Q-матрица данной модели имеет вид, показанный на рисунке
1.9.
Эта модель быстро стала усложняться, так какначало увеличиваться
количество расчетных параметров в Q-матрице. Появились другие модели
замен, такие как K80 (Kimura 2-parameter, Kimura, 1980), F81 (Felsenstein,
1981), HKY85 (Hasegawa, Kishino and Yano, 1985), T92 (Tamura 1992), T93 (Tamura
and Nei, 1993) и GTR: Generalised time-reversible (Tavaré,
1986)[169-172].
модель является наиболее сложной моделью нуклеотидных замен и включает 8
свободных расчетных параметров [173, 174]. В этой модели каждый нуклеотид имеет
свою собственную частоту замен: πA + πG + πT +
πC = 1 и различную
скорость замены основания i на основание j: a+b+c+d+e+f=1(рисунок
1.10).
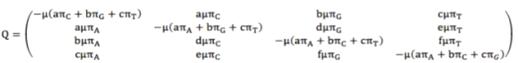
Промежуточные модели, такие как HKY85, рассчитывают индивидуальную частоту появления каждого нуклеотида и различные скорости замены нуклеотидов для транзиций (А → G, C → T) и трансверсий (А → С, G → T). Предположение, что частота нуклеотидных замен одинакова на протяжении всего сиквенса и в каждом сайте с равной вероятностью может произойти мутация, с биологической точки зрения часто не верно. Например, хорошо известно, что третья позиция в кодоне, кодирующем аминокислоту, изменяется чаще, чем первая и вторая позиции кодона. Чтобы учесть этот факт, обычно используется модель нуклеотидных замен, позволяющая рассчитать гетерогенность между сайтами и предполагать, что некоторые сайты являются инвариантными. Было обнаружено, что расчет гетерогенности сайтов хорошо описывается гамма-распределением (G), при котором каждый сайт имеет свою степень изменения нуклеотидов.
Модели реконструкции филогенетических деревьев
Следующим шагом определения филогенетических отношений является выбор соответствующей модели реконструкции филогенетического дерева. Наиболее ранние и простые модели - дистанционные, в основном модель «минимальной эволюции» (minimum evolution, ME) и кластерный анализ. Обе модели реконструируют дерево, используя матрицу попарных генетических дистанций. Методы кластеризации, как, например, UPGMA (Unweighted Pair Group Method with Arithmetic Mean), предполагают постоянную скорость эволюции во времени и реконструируют деревья, поэтапно группируя сиквенсы с наименьшей дистанцией между ними. Сиквенсы кластрируются, основываясь на усреднении расстояний, то есть глубина дивергенции - это дистанция между сиквенсами А и В, деленная на 2. Методы, рассчитывающие минимальную эволюцию, как, например, neighbor-joining (NJ), сходны с методами кластерного анализа, но они не предполагают постоянную скорость эволюции всей «родословной» филогенетического дерева. Более того, методы минимальной эволюции основываются на данных, обратных средним дистанциям. Так, дистанция между двумя сиквенсами равна сумме длин ветвей между ними, а не средним дистанциям. Метод NJ работает следующим образом: он конвертирует алаймент сиквенсов в матрицу дистанций, которая отображает расчетную эволюционную дистанцию между всеми сиквенсами в алайменте. Когда матрица построена, алгоритм находит пару таксонов с наибольшей схожестью и соединяет их с так называемым наиболее близким общим предком (MRCA, most resent common ancestor), представляющим собой узел, соединяющий эти два таксона. Далее дистанции от них до MRCA рассчитываются в зависимости от выбранной модели нуклеотидных замен и, в конечном итоге, определяются расстояния от MRCA до всех таксонов в алайменте. В результате образуется новая матрица, где два первых таксона представляют один наиболее близкий общий предок, MRCA и алгоритм начинает заново рассчитывать расстояния до оставшихся таксонов. Этот процесс повторяется до тех пор, пока все таксоны не будут включены в филогенетическое дерево.
Метод максимального правдоподобия (Maximum Likelihood, ML) позволяет вычислить условную вероятность параметров относительно данных и модели [175]. Метод заключается в следующем: на основе выборки данных и предполагаемой модели (нормальное распределение) алгоритм случайным образом предлагает значения параметров (среднего значения и дисперсии). Затем эти два значения параметров, а также сами наблюдаемые данные подставляются в формулу функции правдоподобия. После расчета получается число - значение правдоподобности. Затем алгоритм ML предлагает новые два значения среднего и дисперсии (или одно из них), опять вычисляет правдоподобность и проверяется, как изменилось ее значение - если оно возросло, алгоритм считает, что он движется в правильном направлении, и предложенные новые параметры более точно описывают данные. Полученные значения принимаются как текущие, и алгоритм продолжает вычисления. Если же значение правдоподобности уменьшилось, то параметры эти считаются неверными, и алгоритм продолжает поиск от тех параметров, что были до этого. Такой поиск идет до тех пор, пока не будет найден так называемый пик функции правдоподобия - такое ее состояние, когда изменение параметров в любом направлении будет приводить к уменьшению значения правдоподобности. Однако число возможных деревьев значительно возрастает с ростом числа таксонов в алайменте, и при их количестве более 10 осуществить поиск всех вероятных деревьев целиком - задача практически невозможная из-за лимита вычислительных возможностей.Для решения этой проблемы в ML были предложены несколько стратегий, которые исследуют части филогенетических деревьев. Таковымиявляются SPR - subtree pruning and regrafting, NNI - nearest neighbor interchange и TBR - tree bisection and reconnection. Все три метода используют специальные правила для генерации нового «переделанного» дерева из заданного стартового дерева. Для каждого нового дерева вычисляется значение максимального правдоподобия, и дерево с наибольшим значением используется для повторения процесса.
Оценка достоверности филогенетического анализа
Недостатком обоих моделей NJ и ML является то, что они производят только расчеты филогенетических дистанций, без какого-либо статистического расчета филогенетических связей в дереве. В начале эта проблема решилась путем использования метода, называемого «статистический бутстрэп» (bootstrapping) [176]. Данный метод относится к классу методов генерации повторной выборки. Суть его заключается в многократной генерации серий образцов (псевдорепликатов), в которых заменены местами сайты в алайменте, включая замены (то есть позиция в алайменте может быть использована несколько раз). Псевдорепликаты при этом имеют длину, равную исходному алайменту. Далее с каждым алайментом псевдорепликатов строится филогенетическое дерево и вычисляется количество кластеров с каждым сиквенсом. Значение бутстрэпа показывает, сколько раз сиквенсы в алайменте кластрируются вместе. Например, если алаймент состоит из 4 образцов A, B, C и D, алгоритм бутстрэп построит 4 псевдофилогенетических дерева. И если в 3 из 4 случаев на этих деревьях образцы A и B будут кластрироваться вместе, то значение бутстрэпа для данных образцов составит 75.
Другим классом методов вычисления достоверности филогенетического дерева являются методы, основанные на длине ветви (branch length methods). К ним относятся:
· zero-branch length test (тестнулевойдлиныветви);
· aLRT (approximately likelihood ratio test, тест приблизительного правдоподобия);
· SH-aLRT (Shimodaira-Hasegawa +approximately likelihood ratio test, тест Шимодаира-Хасегава + тест приблизительного правдоподобия).
Среди данных методов наиболее совершенным является SH-aLRT. aLRT очень близок к LRT (тест отношения правдоподобия), используемому для проверки ограничений на параметры статистических моделей, оцененных на основе выборочных данных. aLRT имеет «нулевую гипотезу», то есть тестируемая ветвь филогенетического дерева имеет нулевую длину. Изначально этот тест базировался на критерии хи-квадрат [177]. Внедрение в aLRT теста Шимодариа-Хасегавы (SH) оптимизировало процедуру тестирования статистики «нулевой гипотезы» филогенетической ветви [178]. Кроме этого, SH-aLRT надежный даже для коротких ветвей филогенетического дерева, в отличие от метода бутстрэп [179].
Таким образом, для изучения генетического разнообразия, распространения и молекулярной эпидемиологии ВИЧ филогенетический анализ ВИЧ является наиболее эффективным методом [180, 181].
Анализ литературных данных показывает, что проблема ВИЧ-инфекции остается актуальной. Высокая скорость репликации вируса, низкая точность обратной транскрипции, интегративный механизм взаимодействия с чувствительной клеткой, многократное проникновение разных вариантов вируса в популяцию человекапривели к высокому генетическому разнообразию ВИЧ, что значительно усложнило лечение ВИЧ-инфицированных пациентов и разработку эффективной вакцины.
Определение субтипов ВИЧ, направлений их заноса и распространения на территории страны дает возможность расшифровывать вспышки ВИЧ-инфекции, заражение через кровь и ее продукты, умышленные случаи инфицирования. Рекомбинантные формы ВИЧ представляют отдельную проблему, поскольку наследуют гено- и фенотипические признаки от родительских вариантов ВИЧ различных субтипов. Такие эволюционные события, как рекомбинация, могут привести к появлению вариантов вируса с новыми биологическими свойствами.В этой связи определение субтипов и рекомбинантных форм ВИЧ в разных странах, в том числе и на территории Республики Беларусь, остается весьма значимым и актуальным направлением научных исследований.
Не менее важны контроль за эффективностью лечения ВИЧ-инфицированных пациентов и своевременное определение мутаций резистентности в геноме ВИЧ к препаратам антиретровирусной терапии. Применяемые в настоящее время на территории Республики Беларусь зарубежные тест-системы для генотипирования и выявления мутаций резистентности ВИЧ к антиретровирусным препаратам весьма дорогостоящие, и создание отечественного набора даст возможность сэкономить значительные валютные средства.
Обозначенные проблемы составили предмет нашего научного интереса при
проведении настоящих диссертационных исследований.