Материал: А27631 Бегунов АА методы и средства аналитических измерений
рН-метр (потенциометрический или с нуль-прибором) состоит из цепи, примерно аналогичной примененной в обычном потенциометре, за исключением того, что гальванометр заменен полупроводниковым усилителем. В результате этого при потенциометрическом компенсировании можно легко обнаружить токи менее 10–12 А.
Погрешность pH-метров прямого отсчета обычно составляет до ±0,1 pH, а pH-метры с нуль-прибором обеспечивают погрешность измерения 0,01–0,02 pH, поэтому они предпочтительны для точных измерений.
В практике измерений составляют гальванический элемент из стеклянного измерительного и каломельного сравнительного элект-родов. Для такой системы суммарная ЭДС связана с рН раствора соотношением
Е = Е0 + 0,059 рН, (5.4)
где Е0 определяется составом гальванического элемента. Его значение находят экспериментальным путем для каждой измерительной ячейки с помощью стандартного раствора, называемого буферным.
Если последовательно в одной ячейке измерить значения ЭДС со стандартным буфером Ес и с неизвестным раствором Ех, то на основании предыдущего соотношения получим
pHx= pHc + 16,95 (Ex – Ec). (5.5)
Данное определение принято в качестве инструментального определения рН. Чтобы оно было действительно правильным, значение Е0 должно быть постоянным. Для этого на практике следует подбирать рН стандартного буфера как можно ближе к рН неизвестной пробы. Например, если неизвестный раствор имеет рН, близкий к четырем, то предпочтительно в качестве стандартного буферного раствора использовать бифталат калия (HOOCC6H4COOK) с рН = 4,01 при 25 °С. В конечном счете точность измерения рН определяется постоянством параметра Е0. Для оптимальных условий, когда значения рН стандартного буфера и раствора неизвестной пробы идентичны, погрешность измерения рН неизвестного раствора может составить приблизительно ±0,02 рН.
Стеклянный мембранный электрод обычно менее чувствителен к помехам по сравнению с другими электродами, однако он обладает некоторыми особенностями, которые ограничивают его применение в определенных средах. Так, в растворах, содержащих высокие кон-центрации ионов щелочных металлов, стеклянный мембранный электрод имеет так называемую щелочную погрешность. Эта погрешность возникает из-за того, что катионы, в особенности ион натрия и в меньшей степени ионы лития и калия, конкурируют с ионами водорода за ионообменные центры на поверхности внешнего гидратированного гелевого слоя, в результате чего изменяется потенциал на поверхности раздела фаз на величину, называемую, диффузионным потенциалом.
Значения жидкостных диффузионных потенциалов в зависимости от зарядов, подвижности и концентрации ионов, а также природы растворителя по каждой стороне от поверхности раздела изменяются в широких пределах. Кроме того, во многих гальванических элементах, используемых в анализе, состав растворов по каждую сторону от поверхности раздела жидкостей отличается настолько, что значение жидкостного диффузионного потенциала нельзя предсказать. В лучшем случае, когда два контактирующих слоя довольно схожи по составу и концентрации, жидкостный диффузионный потенциал будет составлять 1–2 мВ, т. е. значение, которое серьезно влияет на правильность результатов, полученных прямым измерением. Часто диффузионный потенциал достигает 10 мВ и более. Его наличие и неизвестность значения жидкостных диффузионных потенциалов являются одними из основных препятствий в использовании прямой потенциометрии для аналитических целей.
Для уменьшения жидкостного диффузионного потенциала до приемлемого уровня между растворами обычно помещают солевой мостик, содержащий насыщенный раствор хлорида калия, потому что ионы калия и хлора имеют близкую подвижность и проявляют тенденцию к мигрированию через жидкостные соединения почти с одинаковыми скоростями. Однако этот прием успешен только тогда, когда концентрации всех других ионных частиц у поверхности раздела растворов малы по сравнению с концентрацией хлорида калия, иначе жидкостные диффузионные потенциалы будут оставаться сравнительно большими.
5.6. Ионометрия
Потенциометрический анализ с использованием ионоселективных электродов (ИСЭ) называется ионометрией. Методы анализа с применением ИСЭ включают в себя прямую потенциометрию, потенциометрическое титрование, метод стандартных добавок, анализ в потоке проб. Ионоселективный электрод – это электрохимический датчик на основе мембраны, потенциал которой служит мерой активности определяемого поля. Мембраны ИСЭ представляют собой растворы электролитов либо твердые или стекловидные электролиты. Разность потенциалов по обе стороны мембраны линейно зависит от логарифма активности определенного ионного компонента в соот-ветствии с уравнением Нернста.
Ионоселективные электроды имеют следующие достоинства:
– они не оказывают воздействия на исследуемый раствор;
– имеют малые размеры;
– пригодны как для прямых определений, так и в качестве индикаторов в титрометрии;
– недороги.
Необходимость анализа биологических объектов, в том числе на клеточном уровне, обусловила важность конструирования и изготовления микроэлектродов, эквивалентные параметры резонансного измерительного контура. Различают три типа ионоселективных мик-роэлектродов (ИСМ):
– стеклянные;
– твердые мембранные ПСМ (для определения хлоридионов);
– жидкостные мембранные (для определения ионов калия, хлора и кальция).
В последнее время интенсивно развивается ионометрия, основанная на использовании ферментов и бактерий, предназначенных для проведения реакций; в результате реакций образуются ионы, к которым чувствителен данный ПСЭ. Дальнейший прогресс в развитии ионометрии связан:
– с разработкой
надежных методов для определения ионов
Na+,
K+,
Ca2+,
CO![]() ,
HCO
,
HCO![]() ,
Cl–;
,
Cl–;
– более широким применением ионоселективных микроэлект-родов.
Разновидностью ионоселективных электродов являются ферментные электроды. В них активный ферментный слой либо непосредственно иммобилизован в ионоселективном электроде, либо располагается в проточной системе. Во втором случае необходимо, чтобы исследуемый раствор вначале соприкасался со слоем иммобилизированного фермента, затем образующийся продукт реакции определяют с помощью проточного ионоселективного электрода. Такое биологическое измерительное устройство называется ферментным реактором. Первым из предложенных и до сих пор наиболее важным потенциометрическим ферментным электродом является электрод, чувствительный к мочевине.
5.7. Основы капиллярного электрофореза
Метод капиллярного электрофореза (КЭФ) появился сравни-тельно недавно. Он основан на разделении компонентов сложной смеси в кварцевом капилляре под действием приложенного элект-рического поля. Микрообъем анализируемого раствора (около 2 нл) вводят в капилляр, предварительно заполненный подходящим буфе-ром – электролитом. При подаче к концам капилляра высокого напря-жения (до 30 кВ) компоненты смеси начинают двигаться по капил-ляру с разной скоростью, зависящей в первую очередь от заряда и массы (точнее – от значения ионного радиуса), в разное время до-стигают зоны детектирования. На полученной электрофорезграмме последовательность пиков фиксирует качественную характеристику вещества (время миграции) и количественную (как в хроматограмме, высота или площадь пика, пропорциональная концентрации ве-щества).
Для более подробного представления о сущности метода рас-смотрим процессы, происходящие в капилляре, заполненном электро-литом и помещенном в продольное электрическое поле.
Плавленый кварц несет на своей поверхности почти исклю-чительно силоксановые группы > Si = O. При контакте с водой они подвергаются гидролизу и образуют силанольные группы > Si, кото-рые затем гидратируются. Скорость и степень гидролиза зависят от температуры и состава водного раствора, в частности от значения рН. Кислотные свойства поверхностных силанольных групп характе-ризуются константой диссоциации: К1 = 4 ·10–3, поэтому при рН > 2,5 на поверхности находятся в большем или меньшем количестве диссоциированные силанольные группы, которые придают ей отри-цательный заряд. При рН < 2 диссоциация силанольных групп прак-тически полностью подавлена и поверхность становится нейтральной.
В большинстве случаев при использовании прямой потен-циометрии для аналитических целей применяют эмпирическую калибровку, которая сводит к минимуму погрешности, возникающие из-за ненадежности определения потенциала электрода сравнения и жидкостных диффузионных потенциалов.
На границе раздела кварц–водный раствор электролита воз-никает так называемый «двойной электрический слой» (ДЭС). Его обкладку составляют отрицательно заряженные гидратированные силанольные группы. В приповерхностном слое электролита к отри-цательно заряженной поверхности кварца примыкают гидратирован-ные катионы, которые образуют вторую обкладку двойного слоя. Из-за мощного электростатического взаимодействия с поверхностью часть катионов, так же как и силанольные группы, частично теряют гидратирующую воду, в результате чего первый слой катионов, непо-средственно контактирующий с поверхностью, становится весьма малоподвижным. Остальная часть нейтрализующих заряд поверхнос-ти катионов распространяется в толщу раствора, образуя так назы-ваемую диффузную часть второй обкладки двойного слоя. Распре-деление катионов между неподвижным и диффузным слоями, а следовательно, и толщина двойного слоя, зависят в первую очередь от общей концентрации электролита в растворе. Чем она больше, тем большая часть положительного заряда диффузного слоя перемеща-ется в неподвижный слой и тем меньшей становится толщина диф-фузного слоя. При концентрации бинарного однозарядного элект-ролита 10–4–10–3 М толщина двойного электрического слоя состав-ляет в среднем 30–50 мкм. Таким образом, при диаметре внутрен-него канала 50–100 мкм практически вся жидкость, заполняющая капилляр, представляет собой диффузную часть двойного электри-ческого слоя.
При наложении электрического поля, направленного вдоль канала капилляра, в нем возникает встречное движение носителей электрических зарядов, в том числе ионов. Так как в диффузной части двойного электрического слоя присутствует некоторая избы-точная концентрация катионов, их движение увлекает за собой (вследствие молекулярного сцепления и трения) всю массу жидкости в капилляре. Возникает так называемый электроосмотический поток (ЭОП), направленный к катоду, который осуществляет пас-сивный перенос раствора внутри капилляра. Скорость ЭОП в сильной степени зависит от рН раствора: в слабокислых растворах она отсут-ствует или незначительна, а в нейтральных и щелочных она воз-растает до максимально возможной. С другой стороны, скорость электроосмотического потока зависит от концентрации электролитов в ведущем буфере: чем больше концентрация электролитов, тем меньшая часть катионов ДЭС остается в диффузионном слое и, соот-ветственно, уменьшается максимально возможная скорость электро-осмотического потока. Наряду с этим под действием электрического поля в капилляре имеют место так называемая «электрическая подвижность ионов» и электрофоретическая подвижность других заряженных частиц. Эта сложная комбинация различных по природе и свойствам процессов, происходящих в капилляре при наложении электрического поля, используемая для аналитических целей; она получила название метода капиллярного электрофореза.
Минимально система, реализующая принципы электрофоре-тического разделения, должна иметь следующие узлы: кварцевый капилляр, источник высокого напряжения, устройство ввода пробы, детектор и систему вывода информации (рис. 5.4).
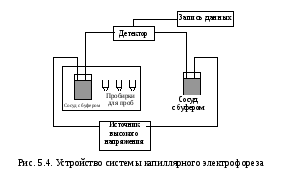
Системы капиллярного электрофореза:
– как правило, используют кварцевые капилляры с внешним полиамидным защитным покрытием: внутренний диаметр 50–75 мкм, внешний – 365 мкм, общая длина 30–100 см;
– для разделения используют положительные и отрицательные напряжения до 30 кВ;
– для ввода пробы применяют избыточное давление (гидроди-намический способ) или высокое напряжение (электрокинетический способ). Объем вводимой в капилляр пробы составляет несколько нанолитров;
– для регистрации электрофореграмм чаще всего используют УФ-детектирование непосредственно в капилляре, в прямом и кос-венном вариантах.
В приборе, реализующем метод капиллярного электрофореза, капилляр, заполненный раствором электролита, своими концами опущен в два содержащих тот же электролит сосуда, в которые вве-дены электроды. Электролит обязательно должен обладать буфер-ными свойствами, чтобы, с одной стороны, воспрепятствовать изме-нению состава раствора в приэлектродных пространствах, а с дру-гой – стабилизировать состояние компонентов пробы в процессе анализа.
Для регистрации сигналов в системе капиллярного электро-фореза используют фотометрическое детектирование с фиксирован-ной или переменной длиной волны.
Дополнительные устройства позволяют автоматизировать по-дачу образцов (автосамплер), осуществлять отвод тепла от капилляра (системы охлаждения капилляра) и управлять прибором, а также собирать и обрабатывать полученные данные с помощью програм-мных продуктов.
При подаче на электроды высокого напряжения в капилляре быстро устанавливается стационарное состояние; при данном состоя-нии через капилляр протекает постоянный электроосмотический по-ток, на который накладывается взаимно противоположная электро-миграция катионов и анионов. Если в капилляр со стороны анода (положительного электрода) ввести небольшой объем раствора пробы, то ЭОП будет переносить эту зону к катоду и зона некоторое время будет находиться в капилляре под воздействием электриче-ского поля высокого напряжения. В течение этого времени компо-ненты пробы, имеющие заряды и отличающиеся от компонентов ведущего электролита, будут перемещаться в соответствии с их электрической подвижностью, специфичной для каждого компонента. Катионные компоненты пробы, двигаясь к катоду, будут обгонять электроосмотический поток. Скорость их движения будет склады-ваться из скорости ЭОП и скорости электромиграции, поэтому на вы-ходе капилляра катионные компоненты будут появляться первыми, и тем раньше, чем больше электрическая подвижность данного иона. Нейтральные компоненты пробы будут перемещаться только под действием ЭОП и появятся на выходе, когда его достигнет зона пробы. Анионные компоненты, перемещаясь к аноду, будут двигаться с меньшими скоростями, чем скорость ЭОП. Некоторые из них, медленно мигрирующие, будут появляться на выходе после выхода ЭОП, а те, чья скорость электромиграции по абсолютной величине превышает скорость ЭОП, вообще будут выходить из капилляра в прианодное пространство.
Если время нахождения пробы в капилляре, которое можно регулировать скоростью ЭОП, напряжением или геометрическими характеристиками капилляра, достаточно, то на выходе капилляра вблизи катода можно фиксировать зоны раствора, в которых нахо-дятся индивидуальные компоненты пробы. Если тем или иным спосо-бом зарегистрировать изменение концентрации компонентов на вы-ходе из капилляра, то полученная запись называется электрофо-реграммой и может служить основой для качественного и количе-ственного анализа смеси.
Вариант зонного капиллярного электрофореза позволяет ана-лизировать компоненты, которые в условиях проведения анализа находятся в форме катионов или анионов. Однако техника капилляр-ного электрофореза может применяться и для анализа нейтральных молекулярных форм веществ вследствие различного распределения аналитов между водным раствором буфера и псевдостационарной мицеллярной фазой. Этот вариант носит название мицеллярной электрокинетической хроматографии (МЭКХ). Данный принцип разделения роднит МЭКХ с обращенно-фазовой ВЭЖХ (ОФ ВЭЖХ).
Традиционно КЭФ сравнивают с ВЭЖХ, поскольку в обоих методах разделение происходит в ограниченном пространстве (ко-лонке или капилляре) с участием движущейся жидкой фазы и для детектирования используются аналогичные принципы. Преимущест-вами КЭФ над ВЭЖХ являются:
– высокая эффективность разделения, недоступная ВЭЖХ и свя-занная с плоским профилем электроосмотического потока;
– малый расход реактивов, при этом практически не требуется применение дорогостоящих высокочистых растворителей: ацетонит-рила, метанола, гексана;
– отсутствие дорогостоящих хроматографических колонок и, следовательно, проблем со «старением» сорбента и заменой коло-нок при выработанном ресурсе;
– отсутствие прецизионных дорогостоящих насосов высокого давления, необходимых для ВЭЖХ;
– простота аппаратурного оформления;
– экспрессность анализа.
Из недостатков КЭФ нужно отметить ограниченное приме-нение метода для образцов, плохо растворяющихся в водных или разбавленных водно-спиртовых растворах; кроме того, невысокую чувствительность при регистрации сигнала в капилляре из-за малой длины оптического пути.
Примером применения КЭФ является анализ консервантов, неорганических катионов и анионов.
В безалкогольных и слабоалкогольных напитках в качестве консервирующих добавок используют бензоат натрия и сорбиновую кислоту. В данных продуктах также можно определять аскорбиновую кислоту (антиоксидант и витаминизирующую добавку) и кофеин. Анионы бензойной, сорбиновой и аскорбиновой кислот определя-ются практически в тех же условиях, что и в методике поверки, т. е. на приборе положительной полярности в виде анионов, выходящих после системного пика, в боратном буферном растворе при рН = 8,6.