Материал: Гиндуллина Хром-метода анлза
5.5.3. Оценка эффективности, селективности и разделительной
способности хроматографических колонок
Одна из основных задач газовой хроматографии состоит в том, чтобы получить хорошее разделение.
Для оценки хроматографического разделения компонентов пользуются тремя группами критериев.
Первая группа критериев зависит от природы сорбента и сорбата, от температуры колонки и характеризует качество разделения в зависимости от различия адсорбируемости или растворимости разделяемых веществ. К критериям этой группы относятся степень разделения α21, и
критерий селективности жидкой фазы КС .
α 21 = |
VR,2 − V0 |
= |
tR,2 − t0 |
= |
lR,2 − l0 |
(1.12) |
|||||||
V |
− V |
t |
R,1 |
− t |
0 |
l |
R,1 |
− l |
|||||
|
|
|
|
||||||||||
|
R,1 |
0 |
|
|
|
|
|
0 |
|
||||
Если α21 = 1, то вещества не разделяются.
Критерий селективности определяется соотношением:
KC = |
VR,2 |
− VR,1 |
= |
tR,2 |
− tR,1 |
= |
lR,2 |
− lR,1 |
(1.13) |
VR,1 |
|
tR,1 |
|
lR,1 |
+ lR,1 |
||||
|
+ VR,1 |
+ tR,1 |
|
||||||
Для неразделяющихся веществ КС = 0 и стремится к 1 при полном разделении компонентов. Селективность жидкой фазы зависит от размеров колонки, природы газа-носителя, от количества введенной в колонку пробы.
Вторая группа критериев обусловлена кинетическими и диффузионными факторами, которые вызывают размывание хроматографических полос. К этим факторам относятся: размеры колонки, природа газаносителя, скорость потока газа-носителя, температура колонки, количество вводимой в колонку пробы и т.д. Совокупность параметров хроматографического опыта, входящих во вторую группу, от которых так же, как и от селективности, зависит качество разделения, можно назвать общим термином - эффективность.
Эффективность хроматографической колонки выражается числом теоретических тарелок или высотой, эквивалентной теоретической тарелке. Число теоретических тарелок зависит от длины колонки и свойств сорбента. ВЭТТ зависит только от типа сорбента и характера его упаковки в колонке.
31
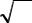
Третья группа критериев учитывает как различие в адсорбируемости или растворимости, так и размывание хроматографических полос. Это так называемые обобщенные критерии. Предложен критерий разделения К, который связан как с селективностью жидкой фазы, характеризующей избирательность, так и с числом теоретических тарелок, характеризующим ее эффективность:
K = 0,212ЧKC |
|
|
N |
(1.14) |
При К = 0,4 достигается достаточно полное разделение. Если требуется лучшее разделение, то при той же скорости потока газа-носителя нужно применить более селективный сорбент.
Вопросы для самоконтроля
1.Что является наиболее важной причиной размывания хроматографического пика?
2.Какая из теорий хроматографии дает основу для оптимизации хроматографического процесса?
3.Нарисуйте зависимость ВЭТТ от скорости потока подвижной фазы в газовой хроматографии?
4.В чем состоит метод теоретических тарелок в хроматографии?
5.Какие основные величины входят в уравнение Ван-Деемтера?
6.Почему в хроматографическую колонку вводят обычно малые количества определяемых соединений?
7.Какие величины характеризуют эффективность хроматографической колонки? Как ее повысить?
8.Какие числовые значения может принимать величина Н? Какое теоретически минимальное значение?
9.Как влияет скорость потока на эффективность хроматографической колонки?
10.Какие хроматографические условия надо менять, чтобы уменьшить вклад в величину Н в уравнении Ван-Деемтера?
5.6. Качественный анализ
Качественными характеристиками хроматографируемых веществ в определенных условиях проведения опыта служат удерживаемый объем и время удерживания. Качественный анализ основан на измерении и сопоставлении этих величин. Существует несколько методов идентификации на основе характеристик удерживания.
1.Применение индивидуальных эталонных веществ. Один из вариантов этого метода состоит в последовательном разделении анализируемой и эталонной смесей в одинаковых условиях. Равенство времен
32
удерживания пиков соответствующих компонентов обеих смесей может служить основанием для идентификации.
Другой вариант заключается в том, что в исследуемую смесь вводят эталонный компонент, наличие которого в этой смеси предполагается. Увеличение высоты соответствующего пика (без его расширения) по сравнению с высотой этого пика на хроматограмме, полученной до введения эталона, может свидетельствовать о присутствии искомого соединения в анализируемой смеси.
Указанный метод прост, но обладает существенными недостатками. Во-первых, необходимо иметь эталонные вещества; во-вторых, все пики, полученные при разделении на данной колонке, должны соответствовать индивидуальным веществам. Но даже при выполнении этих условий нет никаких гарантий однозначности проведенной идентификации. Практически всегда имеются по крайней мере два вещества, удерживаемые объемы которых на колонке с данным сорбентом достаточно близки. Такими веществами вполне могут быть любой компонент смеси
иэталон, нетождественные между собой.
2.Использование табличных данных о характеристиках удерживания. В настоящее время опубликовано много таблиц со значения-
ми относительных удерживаемых объемов для самых различных веществ. Эти таблицы можно использовать при отсутствии необходимых эталонных соединений. Анализируемую смесь разделяют на колонке при условиях, указанных в соответствующей таблице, причем предварительно в смесь вводят небольшое количество веществ, служащих стандартами. На основе полученной хроматограммы рассчитывают относительные удерживаемые объемы, индексы удерживания или другие характеристики. Полученные значения сравнивают с табличными данными.
3. Использование графических или аналитических зависимостей между характеристиками удерживания и другими физико-хи- мическими свойствами веществ. Известно, что логарифм удерживаемого объема, lgVR, в пределахгомологического ряда веществ может коррелировать с такими свойствами, как число углеродных атомов в молекуле (z), температура кипения (Т) и т. д.
lgVR = |
a + bz |
(1.15) |
lgVR = |
a + bT |
(1.16) |
Соответствующие графики широко используют для идентификации компонентов анализируемых смесей. Если заранее известно, к какому гомологическому ряду принадлежит анализируемый компонент, то
33
определенная по графику температура кипения (или число атомов углерода) достаточна для индивидуальной идентификации.
4. Нехроматографические методы идентификации. Эффективным оказалось сочетание газовой хроматографии с другими методами исследования, например, с ИК-спектроскопией и масс-спектрометрией. По каталогу спектров или по эталонным веществам идентифицируют анализируемые вещества.
Возможно использование также методов ядерного магнитного резонанса, пламенной фотометрии и других, включая и химические методы (например, с применением химических реакций до и после хроматографической колонки).
5.7. Количественный анализ
Количественный хроматографический анализ основан на измерении высоты или площади пика, зависящих от концентрации хроматографируемых веществ. Чаще всего для количественных расчетов измеряют площадь пика (S). Для измерения площадей пиков существует несколько приемов. Упрощенный метод состоит в умножении высоты пика (h) на его ширину, измеренную на расстоянии, равном половине высоты (w 1/2 ). Этот метод очень распространен и достаточно точен. Егоприменение возможно при условии получения симметричных пиков и при полном разделении веществ.
Основными методами количественного анализа являются следующие: метод абсолютной градуировки, метод внутреннего стандарта, метод простой нормировки и нормировки с поправочными коэффициентами.
В методе абсолютной градуировки (внешнего стандарта) экспериментально определяют зависимость высоты или площади пика от концентрации вещества и строят градуировочные графики. Далее определяют те же параметры пиков в анализируемой смеси и по градуировочному графику находят концентрацию анализируемого вещества.
Этот простой и точный метод является основным методом определения микропримесей. Кроме того, метод не требует разделения всех компонентов смеси, а ограничивается лишь теми, определение которых необходимо в данном конкретном случае.
Метод внутреннего стандарта основан на введении в анализируемую смесь точно известного количества стандартного вещества. В качестве стандартного выбирают вещество, близкое по физико-химическим свойствам к компонентам смеси. Это вещество должно отсутствовать в исследуемой смеси и давать на хроматограмме пик, отдельный от дру-
34
гих компонентов. После хроматографирования измеряют площади пиков анализируемого компонента (Si) и стандартного вещества (SCT).Массовую долю компонента (Wi, %) рассчитывают по формуле:
ω i = |
Si |
Чr Ч100% |
(1.17), |
|
|||
|
Sст |
|
|
где r – отношение массы внутреннего стандарта к массе пробы. Достоинством метода внутреннего стандарта является хорошая
воспроизводимость, высокая точность, отсутствие влияния на измеряемые величины небольших колебаний условий опыта.
К недостаткам относятся требование точной дозировки стандарта и хорошего отделения пика стандарта от пиков анализируемых веществ. Пользование калибровкой возможно только для той области концентраций, в которой сохраняется линейная зависимость между показаниями детектора и концентрацией определяемого вещества.
Метод простой нормировки чаще всего используют на практике. Для его использования необходимо, чтобы на хроматограмме были зарегистрированы все компоненты, входящие в состав анализируемой смеси; сумму площадей всех пиков принимают за 100 %. Тогда отношение площади одного пика к сумме площадей, умноженное на 100, будет характеризовать массовую долю (%) компонента в смеси.
Этот метод основан на том предположении, что вещества, взятые в одинаковом количестве, дают одну и ту же площадь пика, независимо от их строения. Это приближенно выполняется, если вещества химически сходны, а в качестве газа-носителя применяется газ с высокой теплопроводностью (водород или гелий).
Если чувствительность детектора различна по отношению к разделяемым компонентам смеси, то используют метод нормировки с поправочными коэффициентами. В этом случае расчет ведут по форму-
ле (1.18), где ki – поправочный коэффициент i-го компонента (мг/см2):
ω i = |
ki Si |
|
Ч100 |
|
||
n |
|
(1.18) |
||||
|
е ki Si |
|
||||
|
|
|
||||
|
i= 1 |
|
|
|
|
|
Поправочные коэффициенты получают при анализе стандартных |
||||||
серий и рассчитывают по формуле |
|
|
||||
kiст= |
Sст |
Ч |
ci |
Чk |
(1.19) |
|
|
c |
|||||
|
Siст |
|
|
|||
где С – концентрации определяемого и стандартных веществ. Ме- |
||||||
тод нормировки требует полного |
разделения и |
идентификации всех |
||||
компонентов смеси; необходимости в знании калибровочных коэффици-
35