Материал: Физические основы количественного рентгеноструктурного анализа металлов. Дифрактометрический анализ дефектов кристаллического строения по эффекту уширения линий
Фурье-преобразование соотношения (2) позволяет
получить расчётные ф-лы для вычисления величин Fhkl (в общем случае - комплексных):
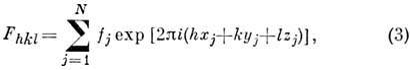
где![]() - ат. фактор рассеяния рентг. излучения атомом jj, xj, yj, zj - его координаты;
суммирование идёт по всем N атомам элементарной ячейки.
- ат. фактор рассеяния рентг. излучения атомом jj, xj, yj, zj - его координаты;
суммирование идёт по всем N атомам элементарной ячейки.
Задача, обратная структурному исследованию,
решается следующим образом: если известна атомная модель структуры, то по (3)
вычисляются модули и фазы структурных амплитуд и, следовательно, интенсивности
дифракционных отражений. Дифракционный эксперимент даёт возможность измерить
мн. сотни не связанных симметрией амплитуд![]() , каждая из которых определяется по (3) набором координат базисных (независимых
по симметрии) атомов структуры. Таких структурных параметров существенно
меньше, чем модулей
, каждая из которых определяется по (3) набором координат базисных (независимых
по симметрии) атомов структуры. Таких структурных параметров существенно
меньше, чем модулей![]() , следовательно,
между последними должны существовать связи. Теория структурного анализа
установила связи разного типа: неравенства, линейные неравенства, структурные
произведения и детерминанты связи структурных амплитуд.
, следовательно,
между последними должны существовать связи. Теория структурного анализа
установила связи разного типа: неравенства, линейные неравенства, структурные
произведения и детерминанты связи структурных амплитуд.
На основе наиболее эффективных статистических
связей развиты прямые методы определения фаз структурных амплитуд. Если взять
тройку больших по модулям структурных амплитуд, индексы которых связаны
простыми соотношениями h1 + h2 + h3 = 0, k1 + k2 + k3 = 0, l1 + l2 + l3 = 0, то
наиб. вероятная сумма фаз этих амплитуд будет равна нулю:
![]()
Вероятность выполнения равенства тем выше, чем больше произведение спец. образом нормированных структурных амплитуд, входящих в это соотношение. С ростом числа атомов N в элементарной ячейке кристалла надёжность соотношения падает. На практике используются существенно более сложные статистические соотношения и достаточно строгие оценки вероятностей выполнения этих соотношений. Вычисления по этим соотношениям весьма громоздки, алгоритмы сложны и реализуются только на мощных современных ЭВМ. Прямые методы дают первые приближённые значения фаз и только наиб. сильных по нормированным модулям структурных амплитуд.
Для практики структурных исследований важны
процедуры автоматического уточнения фаз структурных амплитуд. На основе
приближённого набора фаз![]() сильнейших
структурных амплитуд и по соответствующим экспериментальным модулям
сильнейших
структурных амплитуд и по соответствующим экспериментальным модулям![]() по (2) вычисляется первое приближённое распределение электронной плотности в
кристалле
по (2) вычисляется первое приближённое распределение электронной плотности в
кристалле![]() .
Затем
.
Затем![]() модифицируется на основе физ. и кристаллохим. информации о свойствах этого
распределения. Напр., во всех точках пространства
модифицируется на основе физ. и кристаллохим. информации о свойствах этого
распределения. Напр., во всех точках пространства![]() ,
по модифициров. распределению
,
по модифициров. распределению![]() путём обращения
Фурье вычисляются уточнённые фазы
путём обращения
Фурье вычисляются уточнённые фазы![]() и вместе с экспериментальными значениями
и вместе с экспериментальными значениями![]() используются для построения следующего приближения
используются для построения следующего приближения![]() и т. д. После получения достаточно точных значений
и т. д. После получения достаточно точных значений![]() по
(2) строится трёхмерное распределение электронной плотности в кристалле. Оно по
существу является изображением исследуемой структуры, и вся сложность его
получения вызвана отсутствием собирающих линз для рентг. излучения.
по
(2) строится трёхмерное распределение электронной плотности в кристалле. Оно по
существу является изображением исследуемой структуры, и вся сложность его
получения вызвана отсутствием собирающих линз для рентг. излучения.
Правильность полученной атомной модели проверяют
сравнением экспериментальных![]() и вычисленных
и вычисленных![]() по (3) модулей структурных амплитуд. Количественная характеристика такого
сравнения - фактор расходимости
по (3) модулей структурных амплитуд. Количественная характеристика такого
сравнения - фактор расходимости
Этот фактор даёт возможность методом проб и ошибок получить оптимальные результаты. Для некристаллических объектов это практически единственный метод интерпретации дифракционной картины.
Определение фаз структурных амплитуд прямыми методами осложняется при увеличении числа атомов в элементарной ячейке кристалла. Псевдосимметрия и некоторые др. особенности его строения также ограничивают возможности прямых методов.
Иной подход к определению атомного строения
кристаллов по рентг. дифракционным данным был предложен А. Л. Патерсоном (A. L.
Paterson). Атомная модель структуры строится на основе анализа ф-ции межатомных
векторов P(u,v,w)(ф-ции Патерсона), которая вычисляется по экспериментальным
значениям![]() .
Смысл этой ф-ции можно пояснить с помощью схемы её геом. построения. Атомную
структуру, содержащую в элементарной ячейке N атомов, помещаем параллельно
самой себе так, чтобы первый атом попал в начало координат. Если умножить
атомные веса всех атомов структуры на значение атомного веса первого атома, то
получим веса первых N пиков ф-ции межатомных векторов. Это т. н. изображение
структуры в первом атоме. Затем в начало координат помещаем таким же образом
построенное изображение структуры во втором атоме, затем в третьем и т. д.
Проделав эту процедуру со всеми N атомами структуры, получим N2 пиков ф-ции
Патерсона (рис. 5). Т. к. атомы не являются точками, полученная ф-ция
P(u,v,w)содержит достаточно размытые и перекрывающиеся пики:
.
Смысл этой ф-ции можно пояснить с помощью схемы её геом. построения. Атомную
структуру, содержащую в элементарной ячейке N атомов, помещаем параллельно
самой себе так, чтобы первый атом попал в начало координат. Если умножить
атомные веса всех атомов структуры на значение атомного веса первого атома, то
получим веса первых N пиков ф-ции межатомных векторов. Это т. н. изображение
структуры в первом атоме. Затем в начало координат помещаем таким же образом
построенное изображение структуры во втором атоме, затем в третьем и т. д.
Проделав эту процедуру со всеми N атомами структуры, получим N2 пиков ф-ции
Патерсона (рис. 5). Т. к. атомы не являются точками, полученная ф-ция
P(u,v,w)содержит достаточно размытые и перекрывающиеся пики:
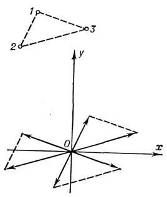
Рис. 5. Схема построения функции межатомных
векторов для структуры, состоящей из трёх атомов.
[![]() - элемент объёма в
окрестности точки (х,у,z)]. Ф-ция межатомных векторов строится по квадратам
модулей экспериментальных структурных амплитуд и является свёрткой
распределения электронной плотности
- элемент объёма в
окрестности точки (х,у,z)]. Ф-ция межатомных векторов строится по квадратам
модулей экспериментальных структурных амплитуд и является свёрткой
распределения электронной плотности![]() с собой, но после инверсии в начале координат.
с собой, но после инверсии в начале координат.
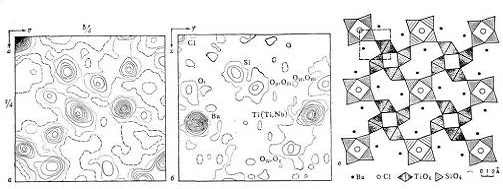
Рис. 6. Минерал баотит
Ba4Ti4(Ti,Nb)4[Si4O12]O16Cl; a - функция межатомных векторов, проекция на
плоскость аb, линии равного уровня значений функции проведены через равные
произвольные интервалы; б - проекция распределения электронной плотности на
плоскость аb, полученная путём интерпретации функции межатомных векторов и
уточнения атомной модели, сгущения линий равного уровня отвечают положениям
атомов в структуре; в - проекция атомной модели структуры на плоскость аb в
полинговских полиэдрах.
Атомы Si расположены внутри тетраэдров из атомов кислорода, атомы Ti и Nb находятся в октаэдрах из атомов кислорода. Тетраэдры [SiO4] и октаэдры [Ti(Nb)O6] в структуре баотита соединены, как показано на рисунке. Атомы Ва и С1 показаны черными и светлыми кружками. Часть элементарной ячейки кристалла, изображённая на рисунках а и б, отвечает на рисунке в квадрату, выделенному штриховыми линиями.
Трудности интерпретации P(u,v,w)связаны с тем,
что среди N2 пиков этой ф-ции необходимо распознать пики одного изображения
структуры. Максимумы ф-ции Патерсона существенно перекрываются, что ещё более
осложняет её анализ. Наиб. прост для анализа случай, когда исследуемая структура
состоит из одного тяжёлого атома и несколько значительно более лёгких атомов. В
этом случае изображение структуры в тяжёлом атоме рельефно выступает на фоне
остальных пиков P(u,v,w). Разработан ряд методов систематич. анализа ф-ции
межатомных векторов. Наиб. эффективными из них являются суперпозиц. методы,
когда две или более копий P(u,v,w) в параллельном положении накладываются друг
на друга с соответствующими смещениями. При этом закономерно совпадающие на
всех копиях пики выделяют одно или несколько из N исходных изображении
структуры. Как правило, для единств. изображения структуры приходится
использовать дополнит. копии P(u,v,w). Проблема сводится к поиску необходимых
взаимных смещений этих копий. После локализации на суперпозиц. синтезе
приближённого распределения атомов в структуре этот синтез может быть
подвергнут обращению Фурье и т. о. он позволяет получить фазы структурных
амплитуд. Последние вместе с экспериментальным значениями![]() используются для построения
используются для построения![]() . Все процедуры
суперпозиции методов алгоритмизированы и реализованы в автоматическом режиме на
ЭВМ. На рис. 6 изображено атомное строение кристалла, установленное
суперпозиционными методами по ф-ции Патерсона.
. Все процедуры
суперпозиции методов алгоритмизированы и реализованы в автоматическом режиме на
ЭВМ. На рис. 6 изображено атомное строение кристалла, установленное
суперпозиционными методами по ф-ции Патерсона.
Разрабатываются экспериментальные методы определения фаз структурных амплитуд. Физ. основой этих методов служит эффект Реннингера - многолучевая рентг. дифракция. При наличии одновременно рентг. дифракционных отражений имеет место перекачка энергии между ними, которая зависит от фазовых соотношений между данными дифракционными пучками. Вся картина изменения интенсивностей при этом ограничена угловыми секундами и для массовых структурных исследований эта методика практического значения пока не приобрела.
В самостоятельный раздел Р. с. а. выделяют
прецизионные структурные исследования кристаллов, позволяющие получать по
дифракционным данным не только модели атомного строения исследуемых соединений,
но и количественные характеристики тепловых колебаний атомов, включая анизотропию
этих колебаний (рис. 7) и их отклонения от гармонического закона, а также
пространственное распределение валентных электронов в кристаллах. Последнее
важно для исследования связи между атомным строением и физ. свойствами
кристаллов. Для прецизионных исследований разрабатываются специальные методы
экспериментальных измерений и обработки дифракционных данных. В этом случае
необходимы учёт одновременных отражений, отклонений от кинематичности
дифракции, принятие во внимание динамических поправок теории дифракции и др.
тонких эффектов взаимодействия излучения с веществом. При уточнении структурных
параметров используют метод наим. квадратов, причём важнейшее значение имеет
учёт корреляции между уточняемыми параметрами.
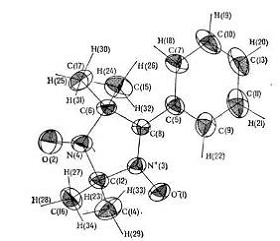
Рисунок 7 - Эллипсоиды анизотропных тепловых колебаний атомов стабильного нитрон-сильного радикала C13H17N2O2.
Р. с. а. используют для установления
связи атомного строения с физ. свойствами сегнетоэлектриков
<#"801805.files/image058.gif"> связана с
энгармонизмом тепловых колебаний атомов Те (рис. 8). Упругие свойства
тетрабората лития Li2B4О7, открывающие для него перспективы применения в
качестве детектора акустических волн, обусловлены характером хим. связей в этом
соединении. С помощью Р. с. а. исследуют распределение в кристалле валентных
электронов, реализующих межатомные связи в нём. Эти связи могут исследоваться с
помощью распределения деформационной электронной плотности, представляющей
собой разность
![]()
где![]() - распределение электронной
плотности в кристалле,
- распределение электронной
плотности в кристалле,![]() - сумма
сферически симметричных плотностей свободных (не вступивших в хим. связи)
атомов данной структуры, которые расположены соответственно в точках с
координатами xi, yi, zi. При установлении по рентг. дифракц. данным деформации
электронной плотности наиб. сложен учёт тепловых колебаний атомов, существ.
образом коррелирующих с характером и направлениями хим. связей. Т. о.,
деформационная плотность
- сумма
сферически симметричных плотностей свободных (не вступивших в хим. связи)
атомов данной структуры, которые расположены соответственно в точках с
координатами xi, yi, zi. При установлении по рентг. дифракц. данным деформации
электронной плотности наиб. сложен учёт тепловых колебаний атомов, существ.
образом коррелирующих с характером и направлениями хим. связей. Т. о.,
деформационная плотность![]() отражает
перераспределение в пространстве той части электронной плотности атомов,
которая непосредственно участвует в образовании хим. связей (рис. 9).
отражает
перераспределение в пространстве той части электронной плотности атомов,
которая непосредственно участвует в образовании хим. связей (рис. 9).
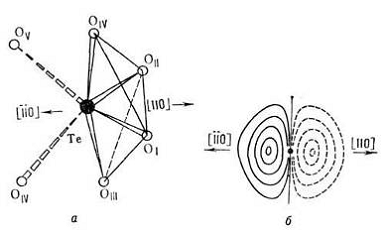
Рис. 8. Ближайшее окружение теллура
атомами О в структуре![]() (a) и ангармоническая
составляющая распределения плотности вероятности нахождения атома Те в данной
точке пространства в процессе тепловых колебаний (б). Положительные (сплошные)
и отрицательные (штриховые) линии равного уровня проведены через 0,02
(a) и ангармоническая
составляющая распределения плотности вероятности нахождения атома Те в данной
точке пространства в процессе тепловых колебаний (б). Положительные (сплошные)
и отрицательные (штриховые) линии равного уровня проведены через 0,02![]() -3
-3
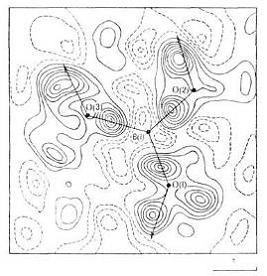
Рис. 9. Сечение синтеза
деформационной электронной плотности кристалла Li2B4O7 плоскостью, проходящей
через атомы О треугольной группы ВО3, в центре которой находится атом В.
Максимумы на отрезках В - О указывают на ковалентный характер связей между
этими атомами. Штриховыми линиями выделены области, из которых электронная
плотность переместилась на химические связи. Линии равного уровня проведены
через 0,2 .![]()
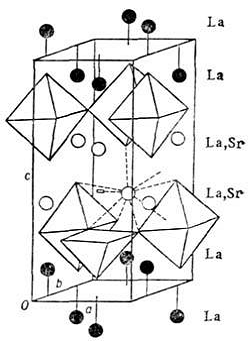
Рис. 10. Упорядоченное размещение
атомов Sr по позициям лантана в структуре![]() Атомы Сu
Атомы Сu
Структурные исследования
высокотемпературных сверхпроводников позволили установить их атомное строение и
его связь с их физ. свойствами. Было показано, что в монокристаллах![]() температура
перехода в сверхпроводящее состояние Тс зависит не только от кол-ва Sr, но и от
способа его статистического размещения. Равномерное распределение атомов Sr в
структуре является оптимальным для сверхпроводящих свойств. Концентрация Sr в
определенных слоях структуры (рис. 10) ведёт к потере в этих слоях части
кислорода и к понижению Тс. Для кристаллов
температура
перехода в сверхпроводящее состояние Тс зависит не только от кол-ва Sr, но и от
способа его статистического размещения. Равномерное распределение атомов Sr в
структуре является оптимальным для сверхпроводящих свойств. Концентрация Sr в
определенных слоях структуры (рис. 10) ведёт к потере в этих слоях части
кислорода и к понижению Тс. Для кристаллов![]() методами Р. с. а. установлено
упорядочение в размещении атомов О. В пределах одного кристалла установлено
наличие ромбических по симметрии областей локального состава
методами Р. с. а. установлено
упорядочение в размещении атомов О. В пределах одного кристалла установлено
наличие ромбических по симметрии областей локального состава![]() с Тс~90 К и
областей находятся в [СuО6]-октаэдрах. Дефектность по кислороду показана
отсутствием у одного из Cu-полиэдров одной кислородной вершины. Позиции,
полностью заселённые атомами La, показаны чёрными кружками. Светлые кружки -
позиции лантана, в которых сконцентрированы и статистически размещены все атомы
Sr.
с Тс~90 К и
областей находятся в [СuО6]-октаэдрах. Дефектность по кислороду показана
отсутствием у одного из Cu-полиэдров одной кислородной вершины. Позиции,
полностью заселённые атомами La, показаны чёрными кружками. Светлые кружки -
позиции лантана, в которых сконцентрированы и статистически размещены все атомы
Sr.
![]() с Тс ~ 60 К. В кристаллах с кол-вом
кислорода меньше чем 6,5 атома на элементарную ячейку, наряду с областями
ромбич. симметрии локального состава
с Тс ~ 60 К. В кристаллах с кол-вом
кислорода меньше чем 6,5 атома на элементарную ячейку, наряду с областями
ромбич. симметрии локального состава![]() появляются области тетрагональной
симметрии локального состава
появляются области тетрагональной
симметрии локального состава![]() , которые не переходят в
сверхпроводящее состояние.
, которые не переходят в
сверхпроводящее состояние.
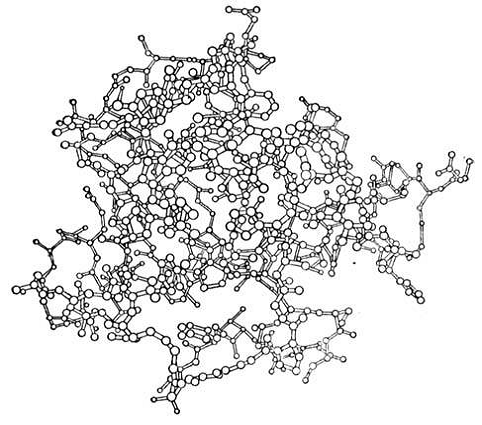
Рис. 11. Атомная модель молекулы
гуанил-специфичной рибонуклеазы С2, построенная на основе рентгеноструктурного
исследования монокристаллов этого белка с разрешением 1,55![]()
Для решения мн. задач физики <#"801805.files/image078.gif">
Рис.10. Точечные дефекты
Вакансия - отсутствие атомов в узлах кристаллической решетки, «дырки», которые образовались в результате различных причин. Образуется при переходе атомов с поверхности в окружающую среду или из узлов решетки на поверхность (границы зерен, пустоты, трещины и т. д.), в результате пластической деформации, при бомбардировке тела атомами или частицами высоких энергий (облучение в циклотроне или нейтронной облучение в ядерном реакторе). Концентрация вакансий в значительной степени определяется температурой тела. Перемещаясь по кристаллу, одиночные вакансии могут встречаться. И объединяться в дивакансии. Скопление многих вакансий может привести к образованию пор и пустот.
Дислоцированный атом - это атом, вышедший из узла решетки и занявший место в междоузлие. Концентрация дислоцированных атомов значительно меньше, чем вакансий, так как для их образования требуются существенные затраты энергии. При этом на месте переместившегося атома образуется вакансия.
Примесные атомы всегда присутствуют в металле, так как практически невозможно выплавить химически чистый металл. Они могут иметь размеры больше или меньше размеров основных атомов и располагаются в узлах решетки или междоузлиях.
Точечные дефекты вызывают незначительные искажения решетки, что может привести к изменению свойств тела (электропроводность, магнитные свойства), их наличие способствует процессам диффузии и протеканию фазовых превращений в твердом состоянии. При перемещении по материалу дефекты могут взаимодействовать.
Линейные дефекты.
Основными линейными дефектами являются дислокации. Априорное представление о дислокациях впервые использовано в 1934 году Орованом и Тейлером при исследовании пластической деформации кристаллических материалов, для объяснения большой разницы между практической и теоретической прочностью металла.
Дислокация - это дефекты кристаллического строения, представляющие собой линии, вдоль и вблизи которых нарушено характерное для кристалла правильное расположение атомных плоскостей.
Простейшие виды дислокаций - краевые и винтовые.
Краевая дислокация представляет собой линию,
вдоль которой обрывается внутри кристалла край “лишней“ полуплоскости (рис.11.)