Материал: MasterPass _ Pharmacology in 7 Days for Medical Students

MECHANISMS OF ACTION
Organic nitrates and nitrites
1Biochemical: When these drugs are taken orally, they are denitrated in the body (especially in the vascular and liver bed of endothelium) in a stepwise fashion. Initially one radical is removed, then another, by glutathione iso-reductase
enzyme. Hydrolysis takes place and metabolites are released.
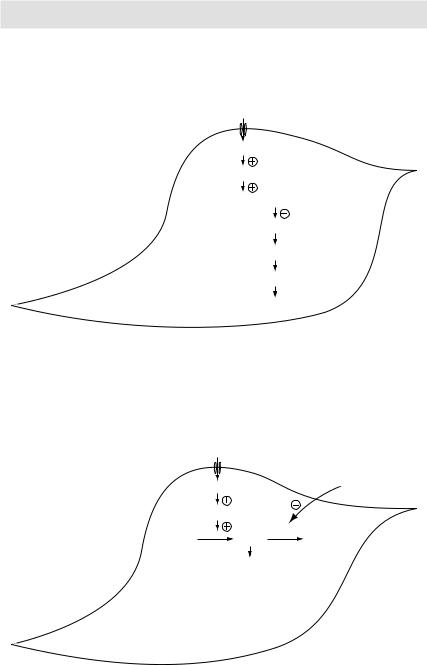
Release of nitric oxide radical → activation of guanylyl (guanylate) cyclase → accumulation of cGMP → activation of cGMP dependent kinases → dephosphorylation of myosin light chain → vasodilatation (venular and arteriolar).
2Hemodynamic: venodilatation → decreased preload → arteriolar dilatation → decreased.
Phosphodiesterase (PDE) inhibitors
Phosphodiesterase (PDE) inhibitors (Sildenafil, tadalafil, vardenafil) fill the penis with blood and are thus used in men with erectile dysfunction. Physiologically, NO released from penile nerve terminals stimulates guanylyl cyclase in the smooth muscle of corpus cavernosum → ↑ intracellular cGMP → smooth muscle relaxation → ↑ inflow of blood → erection. It is pertinent to mention here that if we artificially increase the in vivo NO level by coadministering nitrates and PDE inhibitors, generalised vasodilatation and profound hypotension may result. Thus prescribing PDE inhibitors in patients who are taking nitrates (say due to unstable angina) is contraindicated.
Radioactive iodine I131 therapy
I131 is administered orally in solution form as sodium I131. It is absorbed rapidly, after which it is avidly taken up by the thyroid tissue and incorporated in the storage follicles. I131 destroys thyroidal tissue by the emission of β-rays, which are cytotoxic, ionising and have got short-range (so destroy only follicle cells, not surrounding structures). They have an effective t½ of 5 days and a penetration range of 400–2000μm.
Unlike thioamides and iodine salts, which decrease thyroid hormone synthesis and/or release only transiently, I131 causes complete destruction of thyroid tissue within a few weeks producing permanent hypothyroidism and a cure to thyrotoxicosis without surgery.
Being radioactive, I131 therapy is contraindicated in pregnant ladies and lactating mothers.
Indications of I131 therapy
Thyrotoxicosis due to:
1Graves’ disease.
2Toxic multi-nodular goitre.
3Toxic nodule.
I131 is the only isotope used for the treatment of thyrotoxicosis; others are used in diagnosis. It is the drug of choice for the treatment of Graves’ disease in patients <35yrs of age (except in pregnant/lactating women, in whom it is contraindicated).
Advantages of I131 therapy
1Because of once daily dosage, I131 has good compliance.
2Being odourless, it is not unpleasant to take it.
79
PHARMACOLOGY IN 7 DAYS FOR MEDICAL STUDENTS
Mechanism of vascular smooth muscle relaxation: Nitrates are chemically reduced to release nitric oxide (NO) leading to vasodilatation
Nitric oxide
Nitric oxide
Guanylyl cyclase
GTP  cGMP
cGMP
Myosin phosphatase enzyme
Myosin dephosphorylation
Actin-Myosin crossbridges disengage
Myocyte |
Relaxation |
Figure 3.15 Mechanism of vascular smooth muscle relaxation
Nitric oxide |
|
|
Nitric oxide |
Sildenafil |
|
Guanylyl cyclase |
|
|
GTP |
cGMP PDE |
GMP |
|
Relaxation |
(inactive) |
|
|
|
•PDE degrades cGMP to inactive GMP
•Inhibitors of PDE→↑cGMP→relaxation
Myocyte
Figure 3.16 Mechanism of action of phosphodiesterase (PDE) inhibitors
80

MECHANISMS OF ACTION
3It is cost-effective.
4It is safe (mortality is rare). Fears of radiation-induced genetic damage, leukaemia or neoplasia have not been realised after >50yrs of clinical experience with radioiodine therapy.
5It is a painless procedure, done in an OPD. No hospitalisation of patient is required and thus no work loss is incurred.
6Patient is saved from the hazards of surgery.
Thyroid hormone synthesis and transport
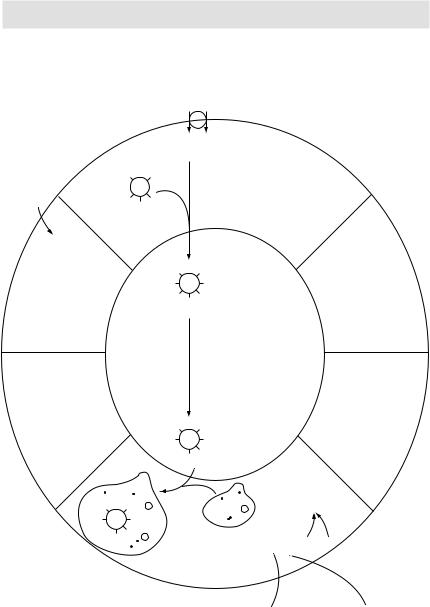
Synthesis of both triiodothyronine (T3) and thyroxine (T4) requires iodine, which is derived from food and iodine supplements (iodine-fortified salt, etc.). Iodine is taken up by the thyroid gland by an active process. Once inside the thyroid gland, the iodide ion combines with tyrosine to form monoiodotyrosine (MIT) and diiodotyrosine (DIT). Next, two molecules of DIT combine to form T4, and one molecule each of MIT and DIT combine to form T3. Once synthesised, these hormones are stored in the thyroid gland. On their release, both T4 and T3 circulate in the blood combined with a protein called thyroxine-binding globulin (synthesised by the liver). In peripheral tissues, some of the T4 is converted to T3 by a process of deiodination. Although T3 is released as such by the thyroid gland also, most of the circulating T3 comes by this process of deiodination of T4 to T3. T3 is about 10 times more potent than T4. It is basically T3 that exerts most of the physiological effects of thyroid hormones at the target organs.
Thyroid-stimulating hormone (TSH) released by the pituitary gland stimulates T4 and T3 synthesis by the thyroid gland. Higher levels of T4 and T3 in turn inhibit pituitary release of TSH thus providing an effective negative feedback control mechanism.
Antithyroid drugs: thioamides
Drugs included: propylthiouracil; carbimazole.
These drugs inhibit the synthesis (not release) of thyroid hormones. They block the process of T4 and T3 synthesis at multiple points:
1They inhibit iodination of the tyrosine residues.
2They block the coupling of MIT and DIT.
3Propylthiouracil alone, when given in high doses, also blocks the peripheral conversion of T4 to T3.
Since these drugs only inhibit the synthesis of thyroid hormones (and not release), their pharmacological effects starts only when the already synthesised and stored thyroid hormone molecules are depleted. This takes about 3–4 weeks.
Iodide salts and iodine
These drugs decrease both the synthesis (by inhibiting the iodination of tyrosine) and release of thyroid hormones. Because of the latter effect, unlike thioamides, these drugs have a rapid onset of action (within 2–7 days). Additionally, these drugs also decrease the size and vascularity of the enlarged hyperplastic thyroid gland. They are thus most beneficial when given in the pre-op period when, by decreasing the vascularity of the thyroid gland, they decrease the chances of per-op and post-op haemorrhage from the thyroid tissue.
The effects of iodide salts and iodine are transient. After a few weeks, the thyroid gland stops responding and escapes from the iodide block rendering these drugs ineffective.
81
PHARMACOLOGY IN 7 DAYS FOR MEDICAL STUDENTS
I– Na+
I– Na+
Thyroid |
T |
TG |
T |
follicular |
|
|
|
cell |
T |
T |
T |
Thyroid peroxidase |
(organification) |
T |
DOT |
|
TG |
MIT MIT MIT |
|
Colloid space
Thyroid peroxidase |
(coupling) |
Phagolysosome
MIT |
|
T4 |
T3 |
TG |
DOT |
|
T |
T |
MIT |
|
T4 |
T3 |
TG |
DOT |
|
T |
T |
Lysosome
I–
 T4, T3, MIT, DIT Proteolysis of
T4, T3, MIT, DIT Proteolysis of
thyroglobulin
Plasma
 Deiodinase
Deiodinase  T4
T4  T3
T3
Peripheral conversion
Figure 3.17 Thyroid hormones synthesis, storage and release
82

MECHANISMS OF ACTION
These drugs are usually given in the form of Lugol’s solution (iodine and potassium iodide).
Aspirin and NSAIDs
As we know that arachidonic acid (derived from the cell membrane lipids) metabolism follows two pathways – lipoxygenase and cyclooxygenase. The latter pathway leads to the formation of three eicosanoids, i.e. prostacyclin, prostaglandins (PGE and PGF) and thromboxane.
Cyclooxygenase (COX) enzyme exists in 2 isoforms – COX-1 and COX-2. COX-1 is found in many body tissues, primarily in the non-inflammatory cells; COX-2, however, is found primarily in the inflammatory cells (polymorphonuclear cells, lymphocytes, etc.).
Aspirin and other non-selective NSAIDs: inhibit both COX-1 and COX-2 in turn inhibiting the synthesis of eicosanoids throughout the body. Eicosanoids are important mediators of inflammation. For example, they are involved in increasing or decreasing vascular and bronchial tone, leukocyte chemotaxis, platelet aggregation, etc. By inhibiting eicosanoids synthesis, these drugs exert their anti-inflammatory effect. For example, PG synthesis in CNS (stimulated by pyrogens) leads to fever. NSAIDs by suppressing PG synthesis, thus produce an antipyretic effect. PGs produced in the injured tissues activate the nociceptors. NSAIDs exert an analgesic effect by suppressing PG synthesis in the injured tissues. PG synthesis in the stomach protects the gastric lining from the cytotoxic effects of HCl. This cytoprotection is lost when PG synthesis is inhibited by the NSAIDs leading to gastritis and peptic ulceration.
Difference between aspirin and other non-selective NSAIDs: Unlike other non-selective NSAIDs, aspirin (but not its active metabolite, salicylate) acetylates and thus irreversibly inhibits COX-1 and COX-2 enzymes. This leads to a longer duration of action (especially the antiplatelet effect).
COX-2 selective NSAIDs (celecoxib and rofecoxib): only inhibit the COX-2 enzyme primarily found in the inflammatory cells. These agents thus do not inhibit eicosanoids synthesis throughout the body. Thus, at least theoretically, these agents should have lesser GI side effects (gastritis and peptic ulceration) – something not uncommonly seen with protracted therapy with aspirin and other non-selective NSAIDs.
GPIIb-IIIa receptor antagonists
Fibrinogen strands bind with GPIIb-IIIa receptors present on platelet membranes leading to platelet aggregation. GPIIb-IIIa receptor antagonists (like tirofiban and abciximab) by inhibiting GPIIb-IIIa receptors inhibit platelet aggregation. Use of these potent antiplatelet drugs is reserved in selected cases of unstable angina and non-ST elevation myocardial infarction (NTEMI) and those undergoing percutaneous coronary intervention.
Clopidogrel, ticlopidine and dipyridamole
cAMP is one of the ‘natural antiplatelets’ found in human bodies. An increase in the intracellular concentration of cAMP in platelets leads to decrease platelet aggregation and vice versa (the exact mechanism not clearly understood). Physiologically, ADP receptor activation causes inactivation of adenylyl cyclase enzyme leading to a fall in cAMP levels. Drugs like clopidogrel and ticlopidine irreversibly inhibit the ADP
83