Материал: MasterPass _ Pharmacology in 7 Days for Medical Students
PHARMACOLOGY IN 7 DAYS FOR MEDICAL STUDENTS
channels in outer membrane of bacteria. It is believed that beta-lactam ring (6-amino- penicillanic acid nucleus) of the bacteria is a structural analogue of D-alanyl-D-alanine. During transpeptidation in the presence of penicillins, beta-lactam ring is incorporated in the cross links rather than D-alanyl-D-alanine and this renders that cross links between peptidoglycan chains weak.
Cephalosporins
1They bind to specific penicillin biding protein (PBP)-receptors on bacteria causing inhibition of cell wall synthesis by blocking transpeptidation of peptidoglycans.
2They also cause activation of autolytic enzymes in the cell wall causing bacterial death.
Sulfonamides
They are bacteriostatic and inhibit protein synthesis. Sulfonamide susceptible organisms, unlike mammals, cannot use exogenous folate but must synthesise it from PABA.
This pathway is essential for the synthesis of purines and nucleic acids. Since sulfonamides are structural analogues of PABA, they inhibit dihydropteroate synthase and thus folate production. Sulfonamides inhibit both gram positive and negative bacteria. Rickettsiae are not inhibited, rather stimulated by sulfonamides. Combination of a sulfonamide with an inhibitor of dihydrofolate reductase (like trimethoprim) provides synergistic activity because of sequential inhibition of folate synthesis.
Tetracyclines
Tetracyclines are bacteriostatic and act by inhibiting bacterial protein synthesis at ribosomal level. They reach their site of action (30S ribosomal subunit of susceptible bacteria) through cell membrane ‘porin’ channels by simple diffusion or by active energy-dependent transport process. They bind reversibly to 30S ribosomal subunit and block biding of amino-acyl tRNA to the receptor site on mRNA-ribosomal complex. Addition of amino acids to the growing peptide chain is prevented thus inhibiting protein synthesis producing bacteriostatic effect.
Tetracyclines also interfere with oxidative phosphorylation, glucose oxidation, bacteria respiration and cell permeability.
Quinolones
They are bactericidal and block bacterial DNA synthesis by inhibiting the following enzymes:
1DNA gyrase (topoisomerase-II).
2Topoisomerase-IV.
Chloramphenicol
It is bacteriostatic for all susceptible microorganisms by protein synthesis inhibition at ribosomal level by binding reversibly to 50S ribosomal subunit near the binding site for macrolides and clindamycin (thus these drugs can interfere with each others actions). Chloramphenicol inhibits binding of amino acids containing aminoacyl tRNA to the acceptor site of the mRNA-ribosomal complex. Interaction between peptidyl transferase and its amino acid substrate is prevented. Amino acids are not added to the growing peptide chain so protein synthesis is inhibited.
74

MECHANISMS OF ACTION
Chloramphenicol may be bactericidal for Haemophilus influenzae, Nisseria meningitidis and some strains of bacteroides.
Amoebicides
Amoebicides irreversibly inhibit protein synthesis by blocking the movement of ribosomes along mRNA. They act only against amoebic trophozoites.
Aminoglycosides
They are bactericidal against aerobic, Gram –ve bacteria and some Gram +ve cocci. Aminoglycosides are irreversible inhibitors of protein synthesis. The initial event is passive diffusion via poring channels across the out membrane. Drug is then actively transported across the inner membrane into the cytoplasm by an oxygen-dependent process. The transmembrane electrochemical gradient supplies the energy for this process and transport is coupled to a proton pump. Low extracellular pH and anaerobic conditions inhibit the transport by reducing the gradient. Transport may be enhanced by drugs which inhibit cell wall synthesis such as β-lactam penicillins or vancomycin. This enhancement may be the basis of synergism of these antibiotics with aminoglycosides. Once inside the cytoplasm, aminoglycosides bind to specific 30S ribosomal subunit proteins (S12 in case of streptomycin). Protein synthesis is inhibited in at least three ways:
1Aminoglycosides distort the structure of 30S ribosomal subunit, thus interfering with the initiation of protein synthesis.
2Misreading of mRNA, which causes incorporation of incorrect amino acids into the peptide resulting in the formation of nonfunctional or toxic proteins.
3Break up of polysomes into nonfunctional monosomes.
These activities occur more or less simultaneously and the overall effect is irreversible and lethal for the cell.
Aminoglycosides also damage inner cytoplasmic membrane by incorporating defective proteins in the cytoplasmic bacterial membrane. The membrane loses its permeability throwing small and large molecules (structural and functional proteins) leading to bacterial cell death. This is called energy-dependent phase-II transport.
Macrolides
At usual doses, macrolides are bacteriostatic; at higher doses, however, they become bactericidal. Their binding site is either the same or in close proximity to that of clindamycin and chloramphenicol. These drugs bind with 50S subunit of the bacterial ribosome thus inhibiting the translocation step of protein synthesis. Additionally, they may also interfere with the transpeptidation step.
Metronidazole
This drug is particularly effective against anaerobic protozoan parasites (including entamoeba histolytica, etc.). These organisms possess electron-transport proteins that participate in electron removal reactions. The nitro group of metronidazole acts as an ‘electron acceptor’ resulting in formation of cytotoxic compounds that bind to proteins and DNA rendering them nonfunctional. This leads to cell death.
75

PHARMACOLOGY IN 7 DAYS FOR MEDICAL STUDENTS
Clonidine
Clonidine, being lipid soluble, crosses the BBB by simple diffusion and is taken up by the adrenergic neurons and the vasomotor centre of the brain. It is an active drug unlike α-methyl dopa, which is a pro-drug. Clonidine acts as an agonist on α2-receptors situated post-synaptically on the neurons. These α-receptors are α2A in nature. When stimulated, they cause increase in the negative feedback on the release of noradrenaline in brain stem resulting in decreased release from centre to periphery mainly in the heart. This causes fall in blood pressure especially in supine and upright positions by causing decrease in the force of contraction and heart rate leading to decrease in cardiac output. Bradycardia is more marked due to stimulation of parasympathetic system by the drug. Fall in blood pressure is accompanied by decrease in the concentration of noradrenaline and renin in the plasma. Postural hypotension is very mild and is seen in hypovolemic patients only. This is because of agonist effect on the peripheral α2B and α1 adrenergic receptors. In smooth muscles of blood vessels, it acts as an agonist causing vasoconstriction.
Alpha-methyl dopa
It is a structural analogue of levodopa. It is usually given orally. Its absorption through GIT is incomplete. The drug crosses the BBB by an active transport process responsible for the transport of aromatic amino acids. After having reached the brain, it is taken up by adrenergic neurons in the brain stem area where it is converted into α-methyl nor-dopamine, which in turn is converted into α-methyl nor-epinephrine. This process of conversion goes on side by side both in brain and in peripheral sympathetic adrenergic neurons. So it is concentrated in the intraneuronal granules in brain and in periphery and is stored there displacing noradrenaline from the storage vesicles. It is released as α-methyl epinephrine both in centre and in periphery. In periphery, it acts as α1 post-synaptic receptor agonist leading to vasoconstriction and increase in blood pressure. It does not act as antihypertensive peripherally. In the brain, α-methyl nor-epinephrine acts as agonist at α2 post-synaptic receptors causing increase in the negative feedback on release of noradrenaline. So less noradrenaline is released resulting in decreased sympathetic flow from centre to periphery. Methyl dopa mainly produces its antihypertensive effects by causing vasodilatation and decreasing total peripheral resistance by causing variably reduction in heart rate and blood pressure.
β2 selective adrenoceptor agonist drugs
These drugs are given by inhalational, oral and parenteral routes. They are β2-selective adrenoceptor agonists and cause bronchodilatation.
Methylxanthines
In high concentrations in vitro, they inhibit several members of phosphodiesterase (PDE) enzyme family. Since phosphodiesterases hydrolyse cyclic nucleotides, this inhibition results in higher concentration of intracellular cAMP and in some tissues cGMP. cAMP is responsible for a myriad of cellular functions including:
1Stimulation of cardiac functions.
2Relaxation of smooth muscles.
3Reduction in the immune and inflammatory activity of specific cells.
In the PDE enzyme family, amongst the various isoforms of phosphodiesterases,
76

MECHANISMS OF ACTION
PDE4 appears to be the most directly involved in the actions of methylxanthines on airway smooth muscle and on inflammatory cells. The inhibition of PDE4 in inflammatory cells reduces their release of cytokines and chemokines, which in turn results in a decrease in immune cell migration and activation.
Another proposed mechanism is inhibition of cell surface receptors for adenosine. These receptors modulate adenylyl cyclase activity, and adenosine has been shown to provoke contraction of isolated airway smooth muscle and histamine release from airway mast cells.
Corticosteroids (as antiasthmatics)
At biochemical level: Corticosteroids decrease the production of pro-inflammatory mediators (like cytokines, leukotrienes, prostaglandins, platelet-activating factor, etc.).
At cellular level: Corticosteroids inhibit the proliferation of T-lymphocytes and are cytotoxic to certain subsets of T-cells (→ ↓ cellular immunity). They also increase the catabolism of IgG (→ ↓ humoral immunity).
In asthmatics, corticosteroids do not directly cause bronchodilatation. In fact, by decreasing the production and release of pro-inflammatory mediators they reduce bronchial reactivity to different allergens and thus cause a reduction in the frequency of asthma exacerbations.
Besides decreasing the production and release of pro-inflammatory mediators, corticosteroids also cause contraction of engorged vessels in the bronchial mucosa. They also potentiate the effects of β- agonists on the airways.
Mast cell stabilisers
Mast cell stabilisers cause alteration in the function of delayed Cl– channels in the cell membrane inhibiting cell activation. This action on airway nerves is thought to be responsible for nedocromil’s inhibition of cough on:
AMast cells for inhibition of early response to antigen challenge.
BEosinophils for inhibition of inflammatory response to inhalation of allergens.
The inhibitory effect on mast cells appears to be specific for cell type since cromolyn has little inhibitory effect on mediator release from human basophils. It may also be specific for different organs, since cromolyn inhibits mast cells degranulation in human and primate lungs but in the skin. This in turn may reflect known differences in mast cells found at different sites as in their neutral protease content.
Leukotriene pathway inhibitors
They either cause inhibition of 5-lipoxygenase thereby preventing leukotriene synthesis or cause inhibition of binding of LTD4 to its receptor on target tissues, thereby preventing its action.
Adrenocorticosteroids
They enter the cell and bind to cytosolic receptors that transport the steroid into the nucleus. The steroid-receptor complex alters gene expression by binding to glucocorticoid response elements (GREs) or mineralocorticoid-specific elements. Tissue specific responses to steroids are made possible by the presence in each tissue of different protein regulators that control the interaction between the hormone receptor complex and particular response element.
77
PHARMACOLOGY IN 7 DAYS FOR MEDICAL STUDENTS
Heparin
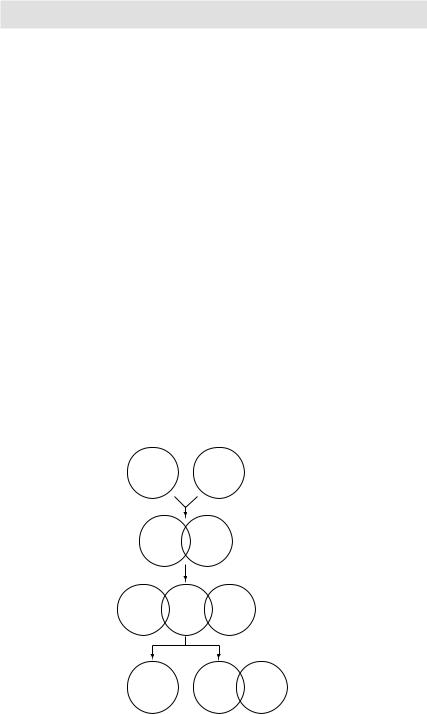
Heparin is a heterogeneous mixture of sulfated mucopolysaccharides. It binds to antithrombin-III (ATIII) forming a heparin-ATIII complex. This complex binds and irreversibly inactivates thrombin (activated factor II), factors IXa, Xa, XIa, XIIa, and XIIIa. In the presence of heparin, ATIII proteolyses clotting factors 1000-fold faster than in its absence.
Warfarin
Warfarin inhibits Vit-K dependent synthesis of factors X, VII, IX and X in the liver by inhibiting the enzyme Vit-K epoxide reductase.
Hormonal contraception (oral/parenteral/implanted)
Combination pills: The combinations of oestrogens and progestins exert their contraceptive effect largely through selective inhibition of pituitary functions that results in inhibition of ovulation. They also produce a change in the cervical mucus, uterine endometrium, motility and secretion in fallopian tubes. All these changes decrease the likelihood of conception and implantation.
Progestins alone pills: Continuous use of progestins alone does not always inhibit ovulation. The other factors mentioned therefore play a major role in the prevention of pregnancy when these agents are used.
Postcoital pills: The exact mechanism of action is not well understood. However, it appears that they inhibit LH surge thus inhibiting ovulation. They probably also produce the same changes in the cervical mucus, uterine endometrium and fallopian tubes as being produced by the combination pills.
Heparin AT-III
Heparin AT-III
Active Heparin AT-III clotting factor
Inactive Heparin AT-III clotting factor
Figure 3.14 Mechanism of action of heparin and antithrombin-III (AT-III)
78