Материал: FTTLabRab
Расшифровка обозначений энергий, вычисляемых при минимизации
Stretch – Энергия вытянутости связей от их оптимальной длины
Bend – Энергия деформации углов между связями от их оптимальной величины
Stretch-Bend – Энергия, необходимая для вытягивания связей, когда угол между ними меньше оптимального
Torsion – Энергия деформации скручивания
Non-1,4 VDW – Энергия пространственного взаимодействия пар атомов, разделенных более чем тремя атомами
1,4 VDW – Энергия пространственного взаимодействия между атомами, разделенными двумя атомами
Dipole/Dipole – Энергия взаимодействия дипольных связей
Total – Алгебраическая сумма энергий.
Heat of Formation – теплота образования связей.
Значение энергий дано в ккал/моль. При этом Chem3D воспринимает любое сочетание атомов на экране, как одну молекулу и вычисляет энергию в расчете на нее.
Химические формулы вводятся в виде текста, индексы располагаются в строке. Например, Al2O3 = Al2O3. Ковалентные связи прочерчиваются мышкой от одного атома к другому после нажатия соответствующей кнопки на экранной панели. Длину и порядок связи можно определить, наведя на нее курсор мышки. Для определения расстояния между любыми атомами выделите (черной окружностью) щелчком левой клавиши мышки первый атом и наведите курсор на второй. Все результаты вычислений можно увидеть, открыв протокол под рамкой рабочего поля.
Практическая часть
-
Смоделируйте молекулу Н2 и ее молекулярную орбиталь, измерьте длину межатомной связи. Молекулярную орбиталь можно изобразить в виде точечной поверхности, либо представить молекулу в модели твердого тела. В первом случае нужно выделить черными окружностями молекулу, войти в меню Object – Dot surfaces и включить Show. Во втором случае нужно войти в меню View – Settings – Model Type и включить модель Space Filling. Здесь можно применить различные варианты окраски, объемной и глубинной растушевки моделей.
-
Разместите на экране семь молекул водорода, запустите программу минимизации энергии ММ2. Методом последовательных приближений добейтесь построения плоской шестигранной фигуры – кристаллического кластера. Отметьте характер и величину энергии взаимодействия, измерьте длину межмолекулярных связей. Схематично зарисуйте кластер в отчет с указанием расстояний. Для упрощения выполнения задачи введите на рабочее поле одну молекулу, разверните ее ось перпендикулярно плоскости экрана, скопируйте в буфер обмена, вставьте из буфера шесть раз. Для вставки новой молекулы нужно отменить выделение старой, иначе старая будет заменяться на новую. Рассмотрите кластер в объемном изображении Space Filling. Включите в меню ММ2 молекулярную динамику, пронаблюдайте за деформацией и разрушением кластера в процессе нагрева, отметьте температуру начала сублимации.
-
Разместите на экране две молекулы HCN, пользуясь исходной моделью Balls & Sticks, запустите программу минимизации энергии в ММ2. Опишите взаимную ориентировку молекул. Отметьте характер и величину энергии взаимодействия. Пользуясь принципом кратчайшей длины связей постарайтесь распознать положение основных диполей, сопоставьте длину межатомных и межмолекулярной связей для одноименных пар атомов C-N. Для наибольшей наглядности разверните оси молекул параллельно плоскости экрана.
-
Разместите на экране молекулу воды, измерьте длину связей водород-кислород и угол между ними. Введите вторую молекулу. Запустите программу минимизации энергии в ММ2. Отметьте характер и величину энергии взаимодействия. Оцените деформацию связей – изменение длин и углов. Опишите взаимную ориентировку молекул. Обратите внимание на механизм и роль водородной связи. Объясните все особенности взаимодействия.
-
Смоделируйте образование молекулы хлора из двух отдельных атомов путем минимизации энергии MOPAC (АМ1), определите длину межатомной связи и ее энергию. По величине энергии сделайте вывод о типе связи.
-
Составьте отчет о выполненной работе. Зарисуйте схематически полученные изображения, укажите найденные параметры, сделайте выводы по каждому пункту задания.
Контрольные вопросы
-
Объясните происхождение молекулярной орбитали.
-
Почему расстояние между атомами в молекуле водорода существенно меньше, чем между молекулами в кластере?
-
Какими параметрами различаются связи в парах C-N и C-H?
-
В чем заключается физический смысл операции минимизации энергии?
-
Почему при взаимодействии двух молекул воды они по-разному деформируются?
-
В ряде случаев энергия Ван-дер-Ваальса имеет положительный знак. Как это объяснить?
-
Какое взаимодействие приводит к более прочной связи – Ван-дер-Ваальса или диполь-дипльное? Как это доказать?
Другой характерной особенностью ковалентной связи является ее направленность. Плотность электронного облака между взаимодействующими атомами значительно больше средней его плотности. Угловые соотношения между связями зависят от числа и типа электронов, участвующих во взаимодействии. Так, например, электронные облака для p-электронов вытянуты в трех взаимно перпендикулярных направлениях и это сказывается на направлении ковалентных связей, в которых участвуют p-электроны.
Следует напомнить, что в большинстве случаев взаимодействия многоэлектронных атомов, внешние электроны находятся в гибридных состояниях. Так, в частности, углерод образуют четыре гибридные s–p-ковалентные связи.
На малых расстояниях становится возможным обменное взаимодействие py и pz-электронов, в результате которого возникают облака повышенной электронной плотности, вытянутые параллельно оси молекулы. Силы притяжения к ним положительных ядер направлены под углом к общей оси, но векторная сумма опять же дает стягивающие силы. Этот вид связи известен как π-связь. Перекрывание волновых функций для σ-связей (при прочих одинаковых условиях) больше, чем для π-связей. Поэтому энергия σ-связей превышает энергию π-связей. Например, для атомов углерода на расстоянии 1,54Ǻ отношение интегралов перекрывания для рσ- и рπ-связей равно 2:1.
В ковалентных связях расположение p-орбиталей может отличаться от ортогонального, характерного для свободных атомов. Деформацию осей обуславливают особенности размещения присоединенных атомов. Так в молекуле метана (предлагается рассмотреть с помощью Chem 3d Pro) атомы водорода располагаются по углам тетраэдра, а углы между связями достигают 109,5. В данном случае атомы водорода находятся в частично ионизированном состоянии и имеют положительный заряд. Электростатические силы отталкивания разводят их до наибольшего удаления друг от друга.
Рис. 5. Модель
кристаллической
структуры алмаза
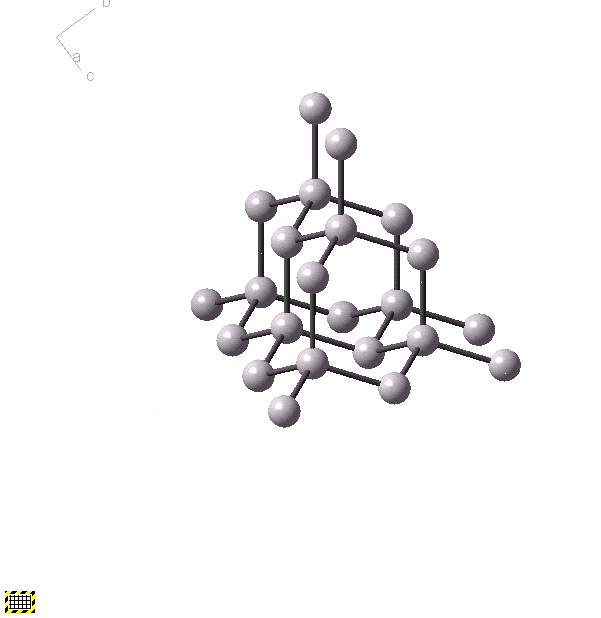
Решетка алмаза кубическая, с периодом a = 3.56 Å, содержит 8 атомов на элементарную ячейку. Атомы первой координационной сферы образуют тетраэдр, аналогичный молекуле метана с углом между связями 109,5. Аналогичные кристаллические решетки с К = 4 имеют другие элементы группы углерода – Si, Ge и Sn (серое) в которых преобладает ковалентная σ-связь. Для определения координационного числа ковалентных кристаллов используют правило K = 8 – N , где 8 – это максимальное число валентных s+p уровней, а N – номер группы элемента в периодической системе Д.И. Менделеева. Таким образом, перечисленные элементы IV группы образуют решетки с К = 4.
Идеальная сбалансированность σ-связи характерна только для алмаза. В других материалах имеет место частичная делокализация валентных электронов. Комбинация σ- и π-связей приводит к возникновению кристаллической структуры иного типа. Так, решетка графита имеет слоистое строение с К = 3. Каждый слой составлен из шестигранников, наподобие молекул бензола, где каждый атом углерода соединен с соседями тремя локализованными σ-связями и ½ π-связи. Здесь π-связи делокализованы, т.к. на 12 связей приходится 6 электронов. Делокализованные электроны слабо связаны с узлами кристаллической решетки и обуславливают электропроводность графита. Межслойная связь осуществляется слабыми межмолекулярными силами.
Решетки с К = 3 образуют элементы V группы, что соответствует правилу K = 8 – N, это P, As, Sb и Bi. В отличие от графита, слои, образованные σ-связями не плоские, а зигзагообразные. В объеме они образуют пакеты, при объединении которых возникает структура ромбоэдрического типа.
Элементы VI и VII групп имеют в соответствии с правилом K = 8 – N по два и одному ближайшему соседу соответственно. В структуре атомы, соединенные ковалентными связями расположены спиральными цепочками. Угол между направлениями связей до ближайших соседей приближается к 90º. Между собой цепочки связаны комбинированной ван-дер-ваальсовой и металлической связью. При объединении цепочек образуется тригональная структура. В частности, в этой структуре кристаллизуется металлический селен. В другой, β-модификации селена цепочки атомов образуют замкнутые кольца. Атомы йода образуют ковалентные молекулы, которые соединяются в ромбоэдрическую структуру силами Ван-дер-Ваальса.
Практическая часть
Для выполнения работы необходимо использовать навыки, полученные в работе №1 и пояснения по работе с пакетом Chem 3d Pro, приведенные там же. Поскольку пакет Chem 3d Pro, главным образом, предназначен для моделирования органических соединений, ковалентные связи со всеми их особенностями использованы как основные. «По умолчанию» задается одинарная связь. Для введения двойной и тройной связи нужно включить соответствующие кнопки.
Минимизацию энергии осуществляйте с помощью приложения МОРАС. Напоминаем, что приложение MOPAC проводит полуэмпирическое моделирование путем решения уравнений Шредингера и содержит 5 потенциальных функций MINDO/3, MNDO, PM3, AM1, и MNDO-d. Всё это методы самостоятельного поля (SCF – Self Consistent Field). Каждая потенциальная функция представляет собой математическое приближение решения уравнения Шредингера для молекул. Для анализа ковалентных связей наиболее эффективен метод АМ1. Для промежуточных построений, в целях экономии времени можно проводить минимизацию энергии в приложении ММ2.
При моделировании кристаллов, как и в случае их реального существования на поверхности возникают оборванные связи, искажающие картину модели. Искажения особенно заметны в случае кристаллов, образованных малым числом атомов. Для снижения краевых искажений рекомендуется оставлять на концах оборванных связей атомы водорода, которые присоединяются программой Chem 3d Pro автоматически. Представьте себе, что Ваш кристалл синтезируется в атмосфере водорода. Реально, в таких условиях имеет место адсорбция водорода на поверхности кристалла; последний всегда будет покрыт моноатомной пленкой из молекул газа. Так что, Ваша модель будет недалека от реалии. А для того, чтобы атомы водорода не затемняли картину, их можно сделать невидимыми, воспользовавшись соответствующей опцией в меню Tools – Show H`s and Lp`s.
Выполните нижеследующие задания.
-
Постройте на экране модель молекулы метана, определите длины связей и углы между ними.
-
Пользуясь схемой на рис.5 постройте модель решетки алмаза. Минимизируйте энергию методом MOPAC (АМ1). Определите углы между направлениями связей и их длину. Путем вращения полученной фигуры найдите положение плоскости [100] и определите период решетки. Сопоставьте полученные данные с табличными для реального кристалла алмаза. Оцените точность моделирования. Необходимо иметь в виду, что правильное измерение кристалло-геометричеких характеристик возможно только для внутренних элементов, где связи полностью уравновешены. Поверхностные атомы находятся в смещенном положении.
-
Постройте модель решетки графита, используя принцип чередования одинарных и двойных ковалентных связей. Определите реальный порядок связи, межатомные и межплоскостные расстояния. Для упрощения задачи постройте одну плоскость из трех-пяти колец, разверните ее горизонтально и перпендикулярно плоскости экрана, скопируйте в буфер обмена, вставьте из буфера два раза, разместите все плоскости одна над другой и запустите программу минимизации в ММ2. Следует напомнить, что Chem 3d Pro не учитывает металлическую связь, поэтому полученная модель качественно и количественно отличаться от решетки реального графита. Найдите эти отличия.
-
Смоделируйте структуры сурьмы (Sb) или висмута, селена (Se) или теллура и йода (I) по принципу комбинации ковалентной и ван-дер-ваальсовой связи, соблюдая правило 8-N. Опишите наблюдаемые особенности. Измерьте характерные межатомные расстояния.
-
Составьте отчет о выполненной работе. Зарисуйте схематически полученные изображения, укажите найденные параметры, сделайте выводы по каждому пункту задания.