Материал: FTTLabRab
The surface that you are plotting is the surface on which square of the orbital you have chosen has a particular value. That value is controlled from the menu for the rendering method you are using. The menu for polygon rendering is found here:
Here is the secondary menu:
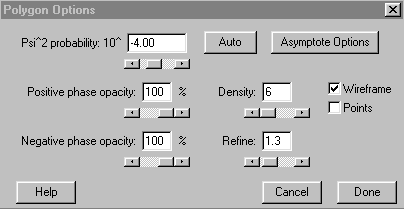
The Psi^2 probability slider allows you to choose the probability value that will be plotted. The Auto button makes a selection for you based on the orbital choice, but you may wish a different value. The other choices on this menu control how the orbital will look. As shown above the orbital would be plotted in wireframe mode. If neither wireframe or points is checked the plot will be a surface with the opacities shown on the left side of the menu.
The information above should be sufficient to enable you to begin to draw and manipulate orbitals. Other menu choices that you may find useful include:
Colors of the background and the orbital phases are controlled from the Colors choice in the File menu.
Orbitals are rotated while holding the left mouse down, and their size is changed while holding the right mouse button down.
Changing the asymptote opacity from 0 adds nodal planes and surfaces to the orbital plot.
Internal nodal structure can be shown by plotting the orbital with a portion cut away (the red partial sphere on the menu bar).
Clicking the menu bar icon that shows an orbital in a box resizes the orbital to fit the screen.
Clicking here opens the Orbital Viewer Manual (requires Adobe Acrobat Reader). The manual is primarily concerned with the methods used by the program to render the orbitals, and may be of limited value to you.
Задания
-
Постройте модель электронной оболочки для атома водорода (Z=1) с различными значениями n, l и m. Постройте аналогичные модели электронной оболочки 1s для атома гелия (Z=2), сравните относительные размеры.
-
Постройте модель электронной оболочки атома бора (Z=5), опишите форму и расположение электронных облаков для различных значений магнитного квантового числа (-1, 0, 1).
-
Определите электронную формулу и постройте модель электронной оболочки атома алюминия (Z=13), опишите форму и расположение электронных облаков. Включите сечение модели для более детального описания модели.
-
Экспериментируйте с различными значениями n, l и m. Для лучшего понимания строения электронных оболочек рекомендуется их зарисовать и описать: сколько наблюдается «лепестков» при заданных значениях n, l и m? Как изменяется вытянутость «лепестков»?
Лабораторная работа №2 моделирование взаимодействия атомов и молекул
Цели работы
1. Ознакомиться с программой химического моделирования Chem 3d Pro.
2. Смоделировать молекулы предложенных веществ, определить особенности и энергию межмолекулярных связей различных типов
Теоретическая часть
Взаимодействие атомов в конденсированном состоянии
Существование определенных межатомных расстояний в твердых телах предполагает равновесие сил притяжения и отталкивания, т.е. химическую связь. Характер связи, ее насыщенность и направленность определяют кристаллическое строение и свойства твердых тел. Как известно из теоретического курса, природа межатомного взаимодействия должна быть электростатической, т.е. можно предположить, что энергия притяжения обратно пропорциональна расстоянию r между атомами (Uпр~ 1/r), но механизм появления сил притяжения и отталкивания требует подробного рассмотрения. Любой отдельный атом представляет собой замкнутую систему с уравновешенными положительными и отрицательными зарядами. В целом атомы электрически нейтральны и, казалось бы, взаимодействовать не должны, но взаимодействуют.
Силы отталкивания, как производная от энергии отталкивания, имеют комплексную природу. Они обусловлены отталкиванием ядер, электронных оболочек, а также специфическими квантово-механическими силами, связанными с расщеплением электронных уровней при взаимодействии атомов. Зависимость энергии отталкивания U от расстояния между атомами может быть установлена эмпирически по объемной сжимаемости кристаллов:
![]() ,
,
где b и n константы, причем n принимает значения от 5 до 11 для различных материалов. Квантово-механический подход приводит к выражению иного типа
![]() ,
,
где аналогично формуле (1) содержатся постоянные b и ρ, но в отличие от (1) для различных материалов ρ отклоняется не более чем на 6 % от своего среднего значения. Обе формулы отражают очень резкое возрастание энергии, а следовательно, и сил отталкивания при сближении атомов.
Рис. 1. Потенциалы сил притяжения,
отталкивания и их сумма
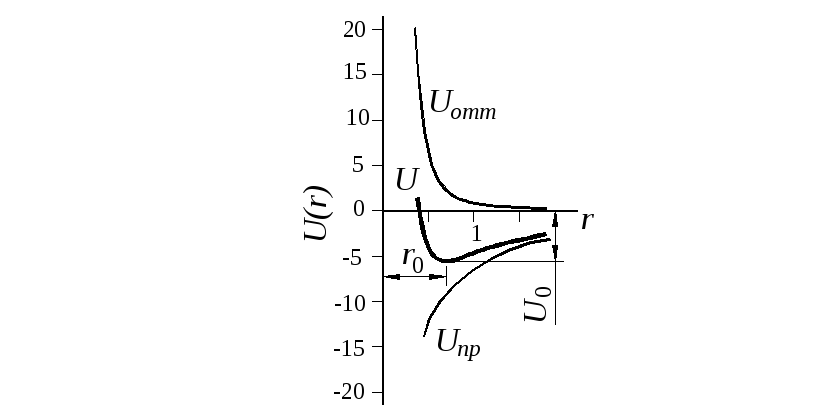
Принцип нахождения минимума энергии (dU/dr = 0) положен в основу моделирования химических связей методами молекулярной динамики.
Молекулярные силы притяжения
Существование жидких и твердых фаз у инертных элементов свидетельствует о существовании слабых сил связи между атомами с застроенными оболочками. Аналогичные силы действуют между молекулами с насыщенными связями. Эти силы называют силами Ван-дер-Ваальса.
а б
Рис. 2. Схема возникновения сил
Ван-дер-Ваальса (а) и схема кристалла,
образовавшегося в результате такого
взаимодействия (б)
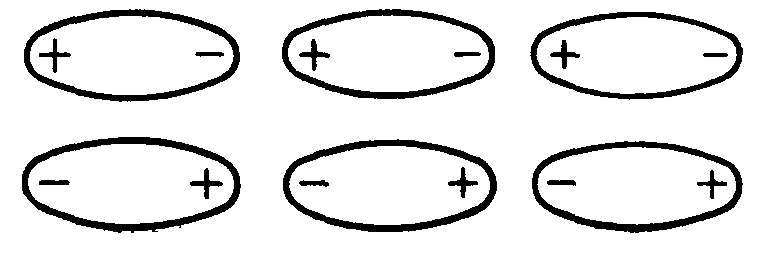

Межмолекулярные связи ненасыщенные и ненаправленные. Это означает, что число ближайших соседей в кристаллической структуре определяется только геометрическими факторами. Таким образом, межмолекулярные силы образую кристаллические решетки координационного типа.
Методическая часть
Назначение и возможности программы Chem 3d Pro
CambridgeSoft Chem3D – программа, разработанная для того, чтобы помочь исследователям в моделировании химических веществ, особенно в области органической химии. Она объединяет в себе мощные возможности построения, анализа и вычисления с дружественным графическим интерфейсом. Программа предоставляет возможности для оптимизации моделей при помощи молекулярной механики, поиска согласования, молекулярной динамики и вычислении энергий молекул.
Chem3D включает в себя инструменты анализа взаимодействия молекул, вставленные в виде приложений. Приложение MM2 позволяет минимизировать энергию методом силового поля Нормана Аллингера (Norman L. Allinger), имитировать динамику молекул при внешнем воздействии и вычислять свойства молекул. Подробное описание этого метода приведено в [1, 2] и на сайте
http://dasher.wustl.edu/tinker
Приложение ММ2 наиболее эффективно для анализа электростатического взаимодействия Ван-дер-Ваальса, дипольного и ионного.
Приложение MOPAC проводит полуэмпирическое моделирование путем решения уравнений Шредингера и содержит 5 потенциальных функций MINDO/3, MNDO, PM3, AM1, и MNDO-d. Все они – методы самостоятельного поля (SCF – Self Consistent Field). Каждая потенциальная функция представляет собой математическое приближение решения уравнения Шредингера для молекул. Исторически эти приближения были сделаны для того, чтобы позволить компьютерам моделировать малые молекулы, хотя большие молекулы все еще эффективнее моделировать при помощи полуэмпирических или молекулярно-механических методов.
Для понимания функционирования потенциальной энергии в полуэмпирической модели MOPAC, приводится хронология приближений, которые включают полуэмпирические методы. Первым приближением было CNDO (Complete Neglect of Differential Overlap – Полное Пренебрежение Дифференциальным Перекрытием). Следующее приближение называлось INDO (Intermediate Neglect of Differential Overlap – Промежуточное Пренебрежение Дифференциальным перекрытием). Затем – MINDO/3, что расшифровывается как "Modified Intermediate Neglect of Differential Overlap" – Модифицированное Промежуточное Пренебрежение Дифференциальным перекрытием. Следующее приближение: MNDO, сокращение от "Modified Neglect of Differential Overlap" (Модифицированное Пренебрежение Дифференциальным перекрытием) что исправило работу MINDO/3 для различных органических молекул, состоящих из элементов первого и второго периодов. AM1 значительно улучшило MNDO. И, наконец, наиболее современный метод PM3 – перепараметризация AM1. Приближение то же, что и для AM1.
Для анализа ковалентных связей наиболее эффективен метод АМ1.
Недостаток пакета Chem 3d Pro в отсутствии модели металлической связи. Это исключает возможность моделирования металлических кристаллов и вносит определенные искажения в модели ковалентных кристаллов, где присутствует металлическая связь, например в моделях решетки графита, сурьмы.
Основные операции и инструменты
|
|
Выделение отдельных атомов и связей. Для выделения нескольких атомов нажмите Shift. Охватывание или Shift + охватывание - выделение областей, содержащих атомы и связи. Для перенесения атома или группы атомов – просто тащите их. С нажатым Alt – перетаскивание вдоль оси Z |
|
|
Вращение всей модели вокруг осей X и Y или вокруг оси Z при нажатом Alt |
|
|
Одиночная ковалентная связь |
|
|
Двойная ковалентная связь |
|
|
Тройная ковалентная связь |
|
|
Некоординированная связь |
|
|
Ввод текста |
|
|
Удаление |
Список литературы
1. Molecular Mechanics, by U. Burkert and N. L. Allinger, ACS, Washington, D.C., USA, 1982.
2. Computational Chemistry, by T. Clark, Wiley, N.Y., USA, 1985.
Лабораторная работа №3 моделирование структуры ковалентных кристаллов
Цели работы
-
Расширить навык атомно-молекулярного моделирования.
-
Получить и проанализировать объемные изображения типичных ковалентных кристаллов, закрепить знания о их строении и об особенностях ковалентной связи
Теоретическая часть
Рассмотрим, каким образом можно описать образование молекулы из двух нейтральных атомов на примере молекулы водорода. Когда атомы водорода находятся далеко друг от друга, их электроны локализованы около ядер, и электронные облака не перекрываются. При сближении атомов происходит перекрытие электронных облаков, и электрон данного атома может перейти к чужому атому. Вероятность такого перехода увеличивается с уменьшением расстояния между атомами. При расстоянии между атомами, равном 2 Ǻ, частота перехода составляет 1014 с–1. При еще большем сближении атомов частота обмена электронами становится настолько большой, что вопрос о том, какому атому принадлежит тот или иной электрон, теряет физический смысл. Электроны коллективизируются, обобществляются, они одновременно принадлежат и тому и другому ядру. Главное условие такого процесса – это наличие свободных энергетических уровней у каждого из атомов.
Электронные облака атомов при подобном взаимодействии образуют единую систему, которая не является простой суммой единичных облаков. На рис. 3 пунктиром (1) показано распределение электронной плотности в свободных атомах водорода; сплошной линией (2) — суммарная плотность электронных облаков при простом их наложении, а линией 3 — действительная плотность электронного облака молекулы водорода. Плотность этого облака между атомами значительно больше суммарной плотности, а вне этого пространства меньше ее. Усиление электронной плотности в пространстве между атомами и приводит к появлению сил притяжения между ними. Дополнительный отрицательный заряд стягивает положительно заряженные ядра, образуя устойчивую систему.
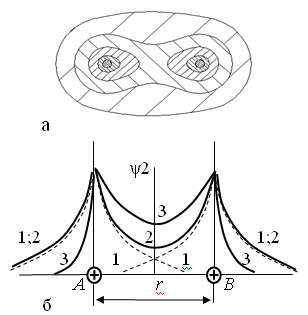
Рис. 3. Распределение электронной плотности в системе из двух атомов водорода
Рис. 4. Схема
образования σ- и π-связей путем попарной
коллективизации валентных электронов:
а – отдельные атомы углерода; б – атомы,
связанные тройной связью (стрелками
показаны силы притяжения)
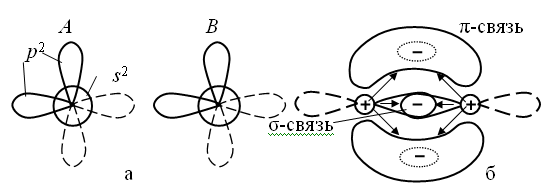
Отличительной особенностью ковалентной связи является ее насыщенность. При образовании молекулы с ковалентной связью стягивается воедино такое число атомов, чтобы коллективизируемые при этом электроны образовали устойчивую оболочку (ns)2(np)6. Поскольку электронное облако ковалентной молекулы полностью застроено, то к данной молекуле не может уже присоединиться ни один атом.