Материал: FTTLabRab
Содержание
ВВЕДЕНИЕ 4
Лабораторная работа №1 6
Компьютерное построение электронных оболочек атомов 6
Теоретическая часть 6
Работа с программой Orbital Viewer 16
Задания 18
Лабораторная работа №2 19
МОДЕЛИРОВАНИЕ ВЗАИМОДЕЙСТВИЯ АТОМОВ И МОЛЕКУЛ 19
Теоретическая часть 19
Методическая часть 21
Практическая часть 24
Контрольные вопросы 26
Список литературы 26
Лабораторная работа №3 26
Теоретическая часть 27
Практическая часть 31
Контрольные вопросы 33
Лабораторная работа №4 34
Теоретическая часть 34
Практическая часть 37
Контрольные вопросы 39
Лабораторная работа №5 39
Построение зоны Бриллюэна для произвольных кристаллических решеток средствами Maple V 39
2 Теоретическая часть 39
3 Выполнение работы 43
Список литературы 50
Введение
Свойства материалов определяются особенностями их кристаллического строения, которое, в свою очередь, зависит от электронного строения атомов, вида и энергии межатомных связей. Особое значение имеют такие понятия, как направленность и насыщенность связей, которые и задают мотив кристаллического строения. Для ясного понимания строения кристаллов важно иметь четкое представление о пространственном расположении атомов и связей между ними. Традиционно в учебной практике эта задача решалась в лучшем случае с помощью моделей, составленных из шаров и проволок, в худшем – с помощью рисунков. Интенсивное развитие компьютерной техники и программного обеспечения предоставило широкие возможности не только для визуализации сложных пространственных структур, но и для вычислительного моделирования межатомного и межмолекулярного взаимодействия, формирующего кристаллические структуры. Компьютерное моделирование заменяет дорогостоящие эксперименты и позволяет выявить недоступные аспекты материального мира. Наибольшее развитие получили численные методы моделирования.
Различные методы моделирования могут быть разделены на четыре основных вида, каждый со своим временным и линейным интервалом. На малых длинах и промежутках времени используются электронно-структурные и квантово-химические методы, которые позволяют получать детальную информацию о малых системах. В симуляциях на уровне атомов основными объектами являются атомы, состояние электронов которых не вычисляется. За счет дальнейшей потери информативности можно значительно увеличить промежутки длины и времени. Значит, становится возможным моделировать зерна и дислокации. В микроструктурных методах основными объектами являются уже элементы микроструктуры, например, сегменты дислокаций или зернограничные элементы. Здесь увеличение отрезков происходит за счет пренебрежения информацией на атомном уровне. Наконец, наибольшие расстояния и промежутки времени – микроны и секунды – охватывают непрерывные методы.
Наиболее конкретное описание свойств тела может быть дано на уровне взаимодействия атомов, но чтобы адекватно перейти к макроскопическим свойствам, нужно описать возможно больший массив взаимодействующих атомов, а это связано с ограничениями возможностей техники.
Широкое развитие получил метод молекулярной динамики, основанный на вычислении сил притяжения и отталкивания и минимизации энергии, которые определяют положение частиц в пространстве относительно друг друга, то есть кристаллическое строение. Результирующая функция имеет ярко выраженный минимум, что соответствует образованию твердой фазы. В простейшем случае алгоритм моделирования основан на интеграции 2-го закона Ньютона по времени. Покажем это на примере алгоритма Верлета.
Закон Ньютона – это Fi = miai , где ai – ускорение атома массы mi. Покажем положение i-го атома во время t-dt и t+dt относительно t:
ri(t+dt) = ri(t) + dt vi(t) + ½ dt2 ai(t) + …
ri(t-dt) = ri(t) - dt vi(t) + ½ dt2 ai(t) - …
Складывая и преобразовывая эти уравнения, получаем:
ri(t+dt) = 2ri(t) - ri(t-dt) + dt2 Fi(t)/mi.
Чтобы в этом алгоритме определить позицию каждого атома во время t+dt, необходимо знать его позиции во время t – dt и t, и силу, действующую на ион во время t.
В научной литературе приводится ряд примеров моделирования на атомном и электронном уровне. В частности, моделирование межатомных связей в кристаллической решетке сложных соединений (например, Al3Li, γ-TiAl, Ti3Al и т. д.), сверхтвердых, магнитных, аморфных, квазикристаллических материалов, изучение взаимодействия дислокаций и точечных дефектов, структуры границ зерен, а также ряд других вопросов. Во многих работах адекватность моделирования доказывается электронно-микроскопическими исследованиями высокого разрешения.
Настоящий цикл лабораторно-практических работ знакомит с методами молекулярного моделирования и визуализации пространственных структур на наиболее простых примерах, требующих минимальных навыков работы с компьютером и начальных знаний в области физики твердого тела. Методом молекулярной динамики моделируются химические связи, рассчитывается их энергия, имитируется формирование молекулярных, ионных и ковалентных кристаллов, моделируются искажения решетки при образовании твердого раствора, вакансии и краевой дислокации. Цель работы №4 – приобретение практических навыков по исследованию процессов теплового расширения, а работа №5 демонстрирует возможности совмещенного термического анализа для изучения фазовых переходов и других процессов, происходящих при нагреве и охлаждении материалов. В целом работы помогают представить необходимые пространственные образы и развивают навыки работы с компьютером и точными физическими приборами, что тоже полезно.
Оформление отчетов
Отчеты по выполненным работам оформляются в соответствии с общими требованиями УГАТУ. Отчет должен содержать название работы, цель, краткие теоретические сведения, методику выполнения, результаты и выводы. Полученные в результате моделирования изображения нужно зарисовать: рисуя, человек лучше понимает и запоминает изображенный предмет. Экспериментальные диаграммы, полученные с помощью копьютеризированных приборов, следует привести в распечатанном виде и выполнить на них разметку характерных точек и участков. Особое внимание уделяется анализу результатов и выводам. Они должны быть по каждому заданию. Чем полнее сделан анализ – тем больше пользы от выполненной работы.
Лабораторная работа №1 Компьютерное построение электронных оболочек атомов
Цель работы: 1. Ознакомиться с программой моделирования электронных оболочек Orbital Viewer;
2. Смоделировать электронные оболочки предложенных атомов, описать форму и расположение электронных облаков.
Теоретическая часть
Наиболее точные представления о состоянии электронов в атоме и конфигурации электронных оболочек, соответствующих каждому состоянию, получены путем решения уравнения Шредингера для атома водорода. Для многоэлектронных атомов доступны лишь приближенные представления, основанные на этих расчетах. Именно поэтому столь важно рассмотреть вопрос о состояниях электрона в атоме водорода.
Электрон в атоме водорода находится в радиально симметричном потенциальном поле, которое создается ядром с положительным зарядом. Потенциальная энергия W электрона описывается формулой
![]() ,
(1)
,
(1)
где r – расстояние электрона от ядра. Таким образом, потенциальная энергия электрона возрастает при удалении от ядра, а потенциальное поле является сферически симметричным. Поэтому уравнение Шредингера для атома водорода удобнее представить в сферической системе координат (рис. 1):
![]() .
.
Решение уравнения Шредингера для атома водорода ищут в виде произведения трех независимых функций:
![]() ,
(2)
,
(2)
где R(r) зависит лишь от r; () зависит только от , а () – лишь от .
Рис. 1. Взаимосвязь между
декартовыми
и сферическими
координатами
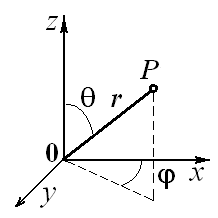
Главное квантовое число п является мерой полной энергии электрона в чисто кулоновском потенциальном поле. Полная энергия электрона в таком поле определяется уравнением:
![]() ,
(3)
,
(3)
где т – масса электрона; е – его заряд, а
![]() ,
,
(h – постоянная Планка).
Главное квантовое число п может быть лишь целым положительным числом, большим нуля: п = 1, 2, 3, 4, ...
Второе квантовое число l, получившее название орбитального (или азимутального), является мерой величины момента количества движения электрона Pl:
![]() .
(4)
.
(4)
Второе квантовое число l не может превышать п–1. Таким образом, каждому значению главного квантового числа п соответствует п значений второго квантового числа: l = 0, 1, 2,..., (n-1). Например, если n = 1, то возможно лишь одно значение l = 0; если же п = 2, то возможны два значения второго квантового числа: l = 0; 1 и т. д.
Третье квантовое число ml, называемое магнитным, характеризует проекцию момента импульса на полярную ось z:
![]() .
(5)
.
(5)
Третье квантовое число может принимать любые целые положительные и отрицательные значения, меньшие по абсолютной величине второго квантового числа l. Величина l определяет момент импульса, a ml – проекцию момента импульса на полярную ось z. Абсолютная величина прямоугольной проекции некоторого вектора не может быть больше величины этого вектора. Отсюда следует
ml l.
Следовательно, третье квантовое число ml может принимать все целые численные значения от –l до +l, включая нуль. Каждому значению l соответствует (2l+1) значений третьего квантового числа ml:
ml = –l, –(l–1), –(l–2), . . ., 0, 1, 2, . . ., l.
Третье квантовое число называют магнитным квантовым числом, так как магнитное поле электрона, вращающегося вокруг неподвижной точки, аналогично магнитному полю тока в кольце. Поэтому магнитное поле вращающегося электрона всегда направлено перпендикулярно плоскости вращения электрона в сторону, определяемую правилом буравчика. Точно так же направлен и момент импульса электрона.
Три квантовых числа появляются в ходе решения уравнения Шредингера для атомов водорода, потому что рассматривается электрон с тремя степенями свободы. Если рассматривать движение электрона в декартовой системе координат, то эти три степени свободы соответствуют перемещению вдоль трех декартовых осей х, у и z.
Электрон обладает четвертой степенью свободы – спином. Спин является особым, сложным свойством электрона (и вообще элементарных частиц), представить которое столь же трудно, как и дуализм «волна – частица». Тем не менее, для большинства физических моделей, использующих понятие спина, вполне достаточным оказывается представление о вращении электрона вокруг собственной оси.
Спин электронов проявляется лишь в магнитном поле. В отсутствие магнитного поля электроны ориентированы беспорядочно, и спиновые свойства электронов в среднем компенсируют одно другое и дают эффект, равный нулю.
Указанные квантовые числа п, l, и ml (табл. 1) позволяют довольно точно описать состояние электронов в атомах и физические явления, связанные с изменением их энергетического состояния.
Энергетическое состояние электронов в атоме определяется, в основном, первыми двумя квантовыми числами. Для характеристики различных энергетических состояний электронов в атоме вводят условное обозначение, состоящее из цифры и буквы. Цифра соответствует значению главного квантового числа. Принятые обозначения второго квантового числа приведены в табл. 2.
В этой сокращенной записи состояние электрона, например, с квантовыми числами п=1 и l=0, можно выразить как 1s; состояние, характеризуемое квантовыми числами п=2 и l= 0, – 2s, состояние с п = 2 и l = 1, – 2р и т. д.
Энергия электрона в атоме водорода определяется только главным квантовым числом п и не зависит от l и ml. Поэтому каждому уровню энергии в атоме водорода соответствует несколько состояний электрона с разными l и ml. Такие энергетически неразличимые состояния называют вырожденными, а число состояний с данной энергией – кратностью вырождения. Так, например, кратность вырождения уровня с n=1 равна 1, уровня с п=2 равна 4, уровня с n=3 равна 9 и т. д.
Таблица 1
Возможные комбинации чисел п, l, и ml
|
n |
l |
ml |
|
1 |
0 |
0 |
|
2 |
0 1 |
0 –1; 0; +1 |
|
3 |
0 1 2 |
0 –1; 0; +1 –2; –1; 0; +1; +2 |
|
n |
0 1 2 … n-1 |
0 –1; 0; +1 –2; –1; 0; +1; +2 … –(n–1); –2; –1; 0; +1; +2…(n–1) |
Таблица 2
Условное обозначение второго квантового числа
|
Значение l |
0 |
1 |
2 |
3 |
4 |
5 |
|
Условное обозначение l |
s |
p |
d |
f |
g |
h |
В результате решения уравнения Шредингера для атома водорода находят также функции R(r), Ф() и (). Произведение этих трех функций определяет волновую функцию, характеризующую состояние электрона в атоме водорода. Волновая функция имеет различный вид для разных энергетических состояний электрона, характеризуемых различными комбинациями квантовых чисел. В частности, для состояния с наименьшей энергией, а именно для состояния 1s, волновая функция имеет вид:
![]() ,
(6)
,
(6)
где а=2/me2=0,529, или в упрощенной записи
![]() .
.
Таким образом, для состояния 1s волновая функция определяется лишь расстоянием от центра атома и не зависит от углов и , т. е. не зависит от направления. Такой класс волновых функций называют сферически симметричными, поскольку значения этой функции во всех точках сферы данного радиуса имеют одинаковое значение. Поскольку 1s изменяется в зависимости от расстояния во всех направлениях совершенно одинаково, то для того чтобы представить себе характер волновой функции 1s, достаточно рассмотреть ее изменение вдоль одного из направлений. Функция 1s монотонно убывает с увеличением расстояния от ядра. Наибольшее значение этой функции соответствует центру атома (r=0).