Материал: Структура и методы синтеза полимеров

Рис.2. Перемещение сегментов макромолекулы под
действием направленных импульсов тепловой энергии: а - случайное перемещение; б
- рептационное (червеобразное) перемещение.
Термодинамическая гибкость характеризует
способность цепи изменять свою конформацию под действием теплового движения и
может быть оценена параметром жесткости, длиной термодинамического сегмента или
параметром гибкости Флори. Чем меньше эти показатели, тем выше вероятность
перехода макромолекулы из одной конформации в другую (табл.4). Параметр жесткости
оценивают отношением среднеквадратичных расстояний между концами реальной и
свободно-сочлененной цепей в разбавленных растворах полимера. Длина
термодинамического сегмента А (сегмента Куна) характеризует такую
последовательность звеньев, при которой каждое звено ведет себя независимо от
других, и также связана со среднеквадратичным расстоянием между концами цепи.
Она равна гидродинамической длине макромолекулы для предельно жестких и длине
повторяющегося звена для предельно гибких цепей. Полимеры диенового ряда и со
связями ~Si-O~
или ~C-O~
в основной цепи характеризуются большей гибкостью по сравнению с полимерами
винилового ряда, так как у них из-за уменьшения обменных взаимодействий между
СН2-группами в 100 раз ниже энергия поворотных изомеров. Природа заместителей
мало влияет на гибкость макромолекул. Параметр гибкости Флори fо
показывает содержание гибких связей в макромолекуле и служит критерием
гибкости, по которому полимеры делят на гибкоцепные (fо>0,63;
А<10нм) и жесткоцепные (fо<0,63;
А>35нм). Последние не бывают в конформации макромолекулярного клубка и имеют
вытянутую форму макромолекул - упругой струны (полиалкилизоцианат, А=100),
коленча-того вала (поли-п-бензамид, А=210) или спирали (биополимеры, А=240).
Таблица 4.
Показатели термодинамической гибкости полимеров в конформации макромолекулярного клубка:
|
Полимер |
Параметр жесткости σж |
Сегмент Куна А, нм |
Число звеньев в сегменте |
|
Сложный полиэфир |
1,3-1,8 |
1 |
- |
|
Полиамид |
1,65-1,85 |
1,66 |
6,6 |
|
Полидиметилсилоксан |
1,4-1,6 |
1,4 |
4,9 |
|
Полидиметилфенилсилоксан |
1,5 |
1,4 |
4,9 |
|
цис-1,4-Полибутадиен |
1,7 |
1,39 |
2-3 |
|
цис-1,4-Полиизопрен |
1,7 |
1,39 |
2-3 |
|
транс-1,4-Полихлоропрен |
1,4-1,67 |
1,39 |
2-3 |
|
Полиэтилен |
2,2-2,4 |
2,08 |
8,3 |
|
Полипропилен |
2,4 |
2,17 |
8,6 |
|
Полиизобутилен |
2,2 |
1,83 |
7,3 |
|
Полистирол |
2,2-2,4 |
2,00 |
7,9 |
|
Поливинилхлорид |
2,8 |
2,96 |
11,7 |
|
Полиакрилонитрил |
2,6-3,2 |
3,17 |
12,6 |
|
Полиметилметакрилат |
1,8-2,2 |
1,51 |
6,0 |
|
Полиоктилметакрилат |
2,3 |
2,0 |
7,0 |
|
Этилцеллюлоза |
4 |
20,0 |
20 |
Кинетическая гибкость макромолекулы отражает
скорость ее перехода в силовом поле из одной конформации в другую и
определяется величиной кинетического сегмента, т.е. той части макромолекулы,
которая отзывается на внешнее воздействие как единое целое. В отличие от
термодинамического сегмента, он определяется температурой и скоростью внешнего
воздействия. С повышением температуры растут кинетическая энергия и гибкость
макромолекулы и уменьшается величина кинетического сегмента. В условиях, когда
время действия силы больше, чем время перехода из одной конформации в другую,
кинетическая гибкость высока, а кинетический сегмент по величине приближается к
термодинамическому сегменту. При быстрой деформации кинетический сегмент близок
к гидродинамической длине макромолекулы, и даже термодинамически гибкая цепь
ведет себя как жесткая. Кинетическая гибкость изолированной макромолекулы
определяется по вязкоупругим свойствам сильно разбавленных растворов с
последующей их экстраполяцией к нулевой коцентрации. Макромолекулы гибкоцепного
аморфного полимера имеют клубкообразную форму как в изолированном виде, так и в
массе. При этом структура полимера не похожа на структуру «молекулярного
войлока», в котором макромолекулы перепутаны хаотически, как считали ранее.
Идея об упорядоченных областях в аморфных полимерах высказана в 1948 г.
Алфреем.
2. Надмолекулярная
структура
При низкой плотности отдельных клубков (0,01-0,03 г/см3) плотность полимеров колеблется в пределах 1-2 г/см3, т.е. в 100 раз больше расчетной, что достигается проникновением клубков друг в друга и параллельностью укладки сегментов в микрообъемах с повышенной плотностью (упорядочения ближнего порядка). Размеры таких микрообъемов колеблются в разных полимерах от 2 до 15 нм. Отношения экспериментальных плотностей кристаллического и стеклообразного полимеров (1,06-1,17) намного выше расчетного показателя (1,54), что указывает на высокую упорядоченность структуры и аморфных полимеров. Возникает своеобразная надмолекулярная структура хаотически перепутанных макромолекул, которые сохраняют форму клубков, а их части (сегменты) образуют микрообъемы упорядоченного взаимного расположения. Формирование даже в аморфном полимере структуры с высокой плотнстью упаковки сегментов и малым свободным объемом обеспечивается высокой гибкостью макромолекул, придающей ему, подобно низкомолекулярной жидкости, небольшую объемную сжимаемость.
Важным признаком надмолекулярной структуры
является связанность всех флуктуаций плотности полимера проходными
макромолекулами (рис.3). Каждая из них проходит через несколько уплотненных
микрообъ-емов, создавая пространственную сетку, узлы которой образованы
доменами ближнего порядка (ассоциатами) и переплетениями (зацеплениями), всегда
возникающими при большой длине макромолекул. Под воздействием теплового
движения и механических напряжений узлы сетки могут распадаться в одном месте и
возникать в другом, поэтому такой тип структуры называют флуктуационной сеткой,
что подчеркивает динамический характер ее возникновения и распада. По
результатам расчета ММ участков проходных макромолекул, соединяющих узлы
флуктуационной сетки, она очень рыхлая: в расплаве полиэтилена - 4.103,
полиизопрена - 10.103 и полистирола - 35.103.
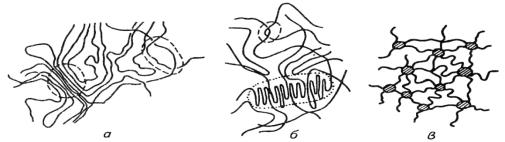
Рис.3. Схематическое изображение возможного
строения флуктуационной сетки и ее узлов: а - ассоциат с параллельной укладкой
сегментов; б - ассоциат складчатый с параллельной укладкой «сложенных»
сегментов (пунктиром выделен узел зацепления); в - схема флуктуационной сетки с
проходными макромолекулами, соединяющими ее узлы.
Цепное строение макромолекул и наличие флуктуационной сетки обуславливают фундаментальную особенность механического поведения полимеров - вязкоэластичность. Деформация в полимере от приложенной внешней силы развивается во времени ее действия. При малом времени (первые доли секунды) перемещаются «свободные» сегменты, не входящие в узлы сетки, что искажает форму макромолекулярных клубков, вытягивает их в направлении действия силы. Поскольку время оседлой жизни «связанных» сегментов (в узлах сетки) больше, узлы не распадаются, и целостность флуктуационной сетки сохраняется. При снятии внешней силы сегменты возвратятся в исходное состояние. Это - обратимая эластическая деформация. При длительном действии нагрузки начнутся распад узлов сетки, перемещение «связанных» сегментов и макромолекул друг относительно друга, что вызовет необратимую вязкую деформацию (течение). Отсутствие четкой границы между окончанием эластической и началом вязкой деформации, одновременное их развитие с преобладанием сначала упругой, а позднее - вязкой и отличают высокоэластическую (вязкоупругую) деформацию, которая может составлять десятки, сотни и даже тысячи процентов. Большие обратимые деформации являются основным признаком полимерной природы материалов, а деформирование - единственным способом оценки их механических свойств.
При охлаждении кристаллизующегося полимера ассоциаты становятся зародышами кристаллизации, которая при температуре плавления происходит медленно, но получаются наиболее совершенные структуры. Только при кристаллизации полиэтилена с ММ около 10000 получены кристаллы с выпрямленными цепями, в которых макромолекулы уложены как карандаши в коробке. Большие гибкие макромолекулы в расплавах полимеров не успевают распрямиться и складываются «гармошкой», создавая многообразие структур. Кристаллит - наименьшее образование из многократно сложенных макромолекул. Монокристалл построен так же, но отличается более высокой степенью упорядоченности и бывает пластинчатым, фибриллярным или глобулярным. Пластинчатые монокристаллы получены для многих полимеров из разбавленных растворов (0,01-0,1%) и состоят из пластинок толщиной 10-26 нм и размерами сторон до 1 мкм которые называют ламелями. Поэтому часто и пластинчатые монокристаллы называют ламелярными.
При неравновесной кристаллизации получают
кристаллы из сложенных цепей. Из кристаллических ламелей выходят петли разной
длины, свободные концы макромолекул и длинные участки, которые могут входить в
структуру соседней ламели. Возникает структура, когда ламели разделены слоями
аморфного полимера и соединены проходными макромолекулами (рис.4). Большая
дефектность отличает такие структуры от монокристаллов.
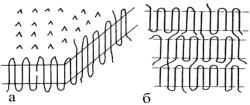
Рис.4. Образование ламели из сложенных цепей (а) и схема
их соединения проходными
макромолекулами в аморфной области (б)
При медленном охлаждении расплавов образуются
пластинчатые и фибриллярные кристаллы больших размеров. Характерная черта
полимеров - возникновение сферолитов - кристаллических образований округлой
формы диаметром от сотен микрон до 1см, которые построены из ламелей, растущих
из единого центра, от одного зародыша кристаллизации, и изгибающихся иногда в
виде спирали. Плоские ламели образуют радиальный сферолит, а спиральные в
полиэтилене высокой плотности - кольцевой сферолит (рис.5).
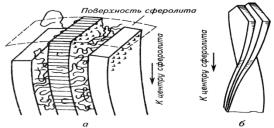
Рис 5. Схема укладки макромолекул и расположение ламелей
в радиальном (а) и кольцевом (б) сферолитах
Таким образом, для кристаллических полимеров
характерен большой набор структурных элементов, характеризующих различные
уровни надмолекулярной организации (рис.6). Полимеры не бывают полностью
закристаллизованы. Соотношение между аморфной и кристаллической фазами
оценивается степенью кристалличности Ко (по объему) или Км (по массе).
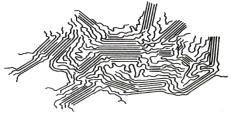
Рис.6. Упрощенная модель возникновения
кристаллитов в массе полимера
Средняя плотность полимера (ρ)
меньше
плотности его кристаллической фазы ρкр,
в которой макромолекулы расположены наиболее упорядоченно, но больше плотноти
аморфной фазы ρам (табл.5).
Степень кристалличности характеризуетет долю полимера в закристаллизованном
состоянии и может быть определена по его плотности: Ко=(ρ-ρам)/(ρкр-ρам);
Км=Коρкр/ρ.
При
комнатной температуре степень кристалличности наиболее распространенных
полимеров лежит в пределах 10-90%, зависит от условий кристаллизации и
повышается с ростом регулярности строения и степени ориентированности
макромолекул. Например, менее разветвленный полиэтилен, полученный при низком
давлении, имеет более высокие значения плотности и степени кристалличности.
Особенности структуры полимеров являются причиной и других особенностей их
кристаллического состояния. Отношение объема, занятого макромолекулами, к
общему объему (коэффициент молекулярной упаковки) у полимерных кристаллов
намного ниже, чем у кристаллов низкомолекулярных веществ, и находится в
пределах 0,65-0,73. Четкая фазовая граница между кристаллической и аморфной
частями отсутствует, а размер кристаллитов мал и на два порядка меньше длины
макромолекул. Кристаллы характеризуются значительной дефектностью и складчатой
структурой, при образовании которой дефекты «выталкиваются» на поверхность
складок, но могут входить и в складчатое образование (ламель). Один и тот же
полимер характеризуется набором кристаллических структур различной морфологии и
различной дефектности. Причины подобной неоднородности кроются, в первую очередь,
в полидисперсности по молекулярной массе, регулярности, конфигурации
макромолекул и т. д.
Таблица 5.
Значения плотностей фаз для некоторых полимеров
|
Полимер |
ρкр, кг/м3 |
ρам, кг/м3 |
ρ, кг/м3 |
|
Полиэтилен высокой плотности |
1000 |
850 |
940-960 |
|
Полиэтилен низкой плотности |
990 |
850 |
920-930 |
|
Полиэтилентерефталат |
1450 |
1330 |
1380 |
|
Поливинилхлорид |
1450 |
1100 |
1140 |
|
Полихлортрифторэтилен |
2190 |
2030 |
2090-2160 |
|
Полиформальдегид |
1500 |
1250 |
1400 |
Рассмотренные выше надмолекулярные структуры формируются при доминирующем влиянии теплового движения. При наложении внешних деформирующих напряжений надмолекулярная структура изменяется, и полимер переходит в ориентированное состояние, при котором оси макромолекул и надмолекулярных образований располагаются вдоль осей ориентации. Ориентированные полимеры широко распространены в природе: волокна хлопка и льна, шелковые нити и шерсть, сухожилия и мышечная ткань. Синтетические ориентированные полимеры можно получить в процессе их синтеза, например полимеризацией в твердой фазе, когда мономер существует в форме монокристалла, или путем их одноосного или двухосного растяжения.
Методы синтеза полимеров играют важную роль в
формировании их структуры. Существуют два основных метода синтеза полимеров -
цепные (полимеризация) и ступенчатые (поликонденсация) реакции. Цепные реакции
реализуются при раскрытии кратных (двойных или тройных) связей или циклов
молекул мономеров и проходят в три стадии: инициирование, рост цепи и обрыв
цепи. Все промышленные мономеры являются производными этилена или бутадиена и
вступают во взаимодействие по механизму цепных реакций, которые подразделяют на
свободнорадикальную или ионную полимеризацию.
3. Свободнорадикальная
полимеризация
Механизм этого метода синтеза был установлен еще
в 30-х годах в работах С.С. Медведева и Г. Штаудингера. Полимеризацию
инициируют свободные радикалы, генерированные тепловым, световым или
радиоактивным воздействиями, которые малоэффективны или сопровождаются
побочными явлениями. Поэтому применяют химические инициаторы (пероксид бензоила,
гидропероксид изопропилбензола, динитрил азоизомасляной кислоты и др.):
(С6Н5СОО)2→2С6Н5СОО*→2С*6Н5+2СО2,
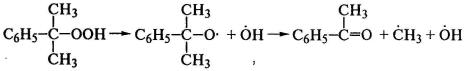 ,
,
![]() .
.
Для ускорения распада инициаторов на радикалы вводят восстановители (амины, сульфиты, тиосульфаты, оксикислоты, соли двухвалентного железа). Окислительно-восстановительные системы снижают энергию активации стадии инициирования со 146 до 50-84 кДж/моль. При распаде гидропероксида в присутствии солей Fe2+ ронгалит (НО-СН2-SO2Na) позволяет легко переводить ионы Fe3+ в Fe2+, и цикл распада инициатора повторяется: