Материал: спектр и фотометр
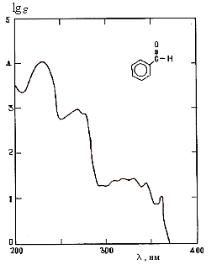
Рис. 10. УФ-спектр бензальдегида
1.3.4. α,β-Непредельные карбоновые кислоты.
В собственно карбоновых кислотах полоса n→π*-перехода карбонильной группы, обладающая малой интенсивностью, расположена в области ~ 200 нм и практически непригодна для идентификации; например, для
уксусной кислоты: СН3СООН λmax (в изооктане) 204 нм (ε 60).
В УФ-спектрах α,β-непредельных кислот проявляется интенсивный максимум π→π*-перехода практически в той же области; например, для акриловой кислоты: СН2=СН-СООН λmax (в изооктане) 200 нм (ε 10000).
Введение алкильной группы в α-положение приводит к батохромному сдвигу максимума на ~ 10 нм, замещение в β-положении ~ 5 нм; наличие пяти- и шестичленного кольца равнозначно двум алкильным заместителям. На этом основании, принимая за исходные данные (основной хромофор) спектр акриловой кислоты, можно с достаточной степенью точности вычислить УФ-спектры простейших α,β-непредельных кислот. Следует отметить, что УФ-спектры этих соединений в значительно меньшей степени, чем спектры соответствующих альдегидов и кетонов, зависят от геометрии молекулы и природы растворителя. УФ-спектры эфиров, амидов, хлорангидридов и ангидридов α,β-непредельных карбоновых кислот практически не отличаются от спектров предельных алифатических кислот.
Таким образом, по данным УФ-спектра может быть достаточно надёжно установлена принадлежность исследуемого вещества к α,β-непре-
26
дельным карбоновым кислотам (или их производным) и сделаны определенные предположения о характере замещения при двойной связи.
1.3.5. Нитрогруппа.
В УФ-спектрах нитросоединений наблюдается интенсивная полоса поглощения, соответствующая переходу π→π* в области, близкой к нижнему пределу измерений (λmax ~ 200 нм, ε 5000), и малоинтенсивная полоса запрещенного n→π*-перехода при 276-280 нм (ε 15-35). Характер заместителя практически не влияет на положение полосы. С увеличением полярности растворителя полоса перехода n→π* смещается в коротковолновую области, как в случае УФ-спектров карбонильных соединений. Поправка, учитывающая влияние применяемого растворителя, составляет (в нм): углеводороды (0), диоксан (-5), этанол (-5), вода (-7).
Таким образом, УФ-спектры нитросоединений относительно мало информативны.
1.3.6. Ароматические гетероциклические системы.
Подобно бензолу, аромати-ческие гетероциклические системы с пятичленным кольцом в УФ-спектре имеют две полосы: интенсивную в коротковолновой области и малоинтенсивную – в длинноволновой; такой же вид имеет УФ-спектр пиридина. Однако при более высокой, по сравнению со спектром бензола, интенсивности длинноволновой полосы её тонкая структура практически исчезает. Переход n→π* в спектрах пятичленных гетероциклов практически не проявляется, так как неподелённая пара электронов гетероатома участвует в образовании ароматического секстета электронов. В УФ-спектре пиридина длинноволновую полосу относят кn→π*-переходу. УФ-спектры основных гетероциклических соединений представлены ниже:
λmax нм (в гексане)
Фуран 200 (ε 10000), 252 (ε1) Тиофен 235 (ε 45000)
Пиррол 210 (ε 15000), 350 (ε 300) Пиридин 195 (ε 7500), 250 (ε 2000)
Изменение положения максимумов при замещении близко к наблюдаемому для бензола.
Таким образом, УФ-спектры ароматических гетероциклических соединений близки по характеру к спектру бензола.
1.3.7. Возможности метода.
Доказательство наличия в исследуемом веществе группировок – хромофоров – сопряжённой диеновой, полиеновой и ароматической систем,
27
а также карбонильной группы или их отсутствия; в простейших случаях возможность определения типа хромофора (табл. 1,2), длины цепи сопряжения, числа алкильных групп при хромофоре; количественный анализ, включая регистрацию изменения концентраций растворов во времени.
1.3.8. Ограничения метода.
Ограниченность рамок применения, так как многие типы органических соединений не имеют максимума поглощения в исследуемой области; сравнительно малые возможности при решении структурно-аналитических задач; в ряде случаев сильное влияние природы растворителя на характер спектра и возможность отклонений от закона Бугера – Ламберта – Бера; фотохимическая изомеризация веществ в процессе работы (например, цuс-транс-изомеризация в диеновых и полиеновых системах).
1.4.ИНФРАКРАСНАЯ (КОЛЕБАТЕЛЬНАЯ) СПЕКТРОСКОПИЯ
1.4.1.Типы колебаний
Энергия, необходимая для возбуждения колебаний атомов в молекуле, соответствует энергии квантов света с длиной волны 1-15 мкм или волновым числом 400-4000 см-1, т. е. электромагнитному излучению средней инфракрасной (ИК) области. Колебательные уровни молекул квантованы, энергия переходов между ними и, следовательно, частоты колебаний могут иметь только строго определенные значения. Поглощая квант света, молекула может переходить на более высокий колебательный уровень, обычно из основного колебательного состояния в возбуждённое. Поглощенная энергия передаётся затем на возбуждение вращательных уровней или преобразуется в кинетическую энергию молекул. Колебания молекул проявляются в двух типах спектров: спектры поглощения в инфракрасной области (ИК-спектры) и спектры комбинационного рассеяния света (спектры КР). Последние в данной работе не рассматривается.
Основными типами колебаний являются валентные и деформаци-
онные.
Валентными колебаниями называются колебания ядер атомов вдоль линии связи, они обозначаются буквой ν(νС = С, ν С=О и т. д.).
Деформационные колебания связаны с изменением валентного угла, образованного связями у общего атома; они обозначаются буквой δ. Для возбуждения деформационных колебаний требуется меньшая энергия, чем в случае валентных колебаний, и, следовательно, они имеют меньшую частоту.
С увеличением числа атомов в молекуле числа возможных колебаний быстро pастёт. В реальной молекуле колебания aтомов тесно связаны
28
друг с другом и взаимодействуют между собой. Спектры молекул представляют собой сложный набор различных колебаний, каждое из которых проявляется в узком интервале частот.
Интенсивность поглощения определяется, как и в УФ-спектроскопии, молярным коэффициентом поглощения, однако в этом случае точность измерения существенна меньше. Обычно интенсивность полос выражают как поглощение (А) или пропускание (Т) светового потока в процентах. Полосы также оценивают по интенсивности как сильные (с.), средние (ср.) и слабые (сл.).
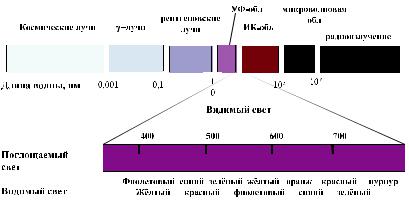
Следует отметить, что инфракрасная область спектра расположена за красным концом видимого спектра (λ>750 нм) (см. рис. 11). Молекулы всех органических соединений поглощают в этой области спектра. Прибор для измерения поглощения в этой области (инфракрасный спектрометр) в принципе не отличается от прибора, используемого для измерения поглощения в видимой и ультрафиолетовой областях спектра (см. рис. 3-7).
Рис. 11. Видимая область составляет только узкую часть спектра электромагнитного излучения
Принципиальные схемы однолучевого и двухлучевого ИК-спектроме- тров приведены на рисунках 12 а и б.
Излучение от источника, дающего непрерывный спектр, проходит через кювету с анализируемым веществом и через кювету сравнения с растворителем в двулучевом приборе и направляется на входную щель монохроматора. После прохождения через дисперги-рующий элемент (призму, дифракционную решетку) монохроматора развернутый в спектр световой поток через выходную щель, сканируясь последовательно по длинам волн, попадает в приемник излучения, который преобразует при-
29
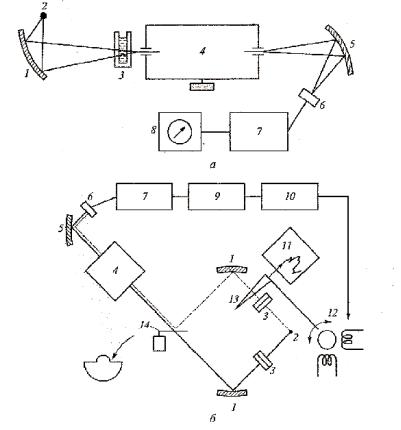
нятый оптический сигнал в электрический и посылает его в систему обработки и измерения. В двулучевом спектрофотометре предусмотрено устройство, позволяющее с помощью перемещаемого фотометрического клина уравнивать световые потоки, идущие через кюветы.
Рис. 12 а и б. Схема ИК-спектрофотометра однолучевого (а) и двухлучевого (б) типа:
1 – зеркало осветителя; 2 – источник излучения сплошного спектра; 3 – абсорбционная кювета; 4 – монохроматор; 5 – сферическое зеркало; 6 – приемник излучения; 7 – усилитель; 8 – измерительная система;
9 – синхронный детектор; 10 – усилитель мощности; 11 – блок регистрации; 12 – сервомотор; 13 – фотометрический клин; 14 – зеркальный обтюратор
Следует отметить, что при поглощении инфракрасного света происходит повышение амплитуды некоторых колебаний в молекуле, и эти
30