Материал: Современное состояние и перспективы использования двойных лекарств в качестве лекарственных средств
Увеличенная ингибирующая активность двухвалентных молекул такрина была
пояснена закреплением к двойному центру. Определение кристаллических структур
ди(5) - такрина и ди(7)-такрина (рисунок 4.1) дало структурное основание для
наблюдения, что ди(7)-такрин - оптимальный ингибитор. Он формирует
благоприятные взаимодействия укладки типа сэндвича в обоих центрах с Trp84 (анионный
центр) и Trp279 (периферический сайт связывания) с минимальной перестройкой
белка. Ди(5)-такрин, который является менее мощным, имеет только одностороннее
благоприятное взаимодействие в периферическом сайте и вызывает конформационные
изменения в белковой цепи возле ацил-связывающего кармана. Структурные
изменения в ферменте, наблюдаемые в комплексе ди(5)-такрин/АХЭ показали, что
выбор оптимальной длины связывающей цепи оказывает существенное влияние на
ингибирующую активность.
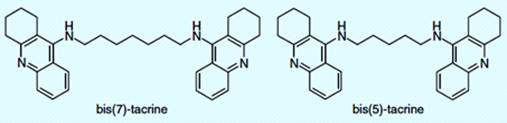
Рисунок 4.1 - Молекулярные структуры сокристаллизующихся ингибиторов АХЭ
Эти результаты подчеркивают проблему текущих, основанных на структуре, подходов дизайна препарата, где не приняты во внимание представления о структуре белка и конформационные изменения. Помимо такрина, также известны другие ингибиторы “анионного центра” АХЭ, которые используются в разработке двойных лекарств АХЭ. Развитие идентичных и неидентичных двойных лекарств контролируется, состыковывая исследования, которые указали на сродство гуперзина и такрина к каталитическим и периферическим сайтам связывания АХЭ.
Исследования моделирования также показали полезные гидрофобные эффекты, переданные алкиленовой связывающей цепью периферическому центру лиганда. Дальнейшее биохимическое исследование АХЭ показало, что периферический сайт связывания в устье ущелья, участвовал в образовании β-амилоидного пептида, отвечающего за нейродегенеративный процесс в наше время. Эта особенность АХЭ инициировала разработку двойного ингибитора сайта связывания, в надежде на увеличение силы ингибирования АХЭ и защиту нейронов от Аβ (amyloid beta) токсичности. Увеличение сродства к АХЭ двойных (-)-галантамина, такрина, гипиридона, производных гуперзина, и гетеродимерных лигандов такрин/пропидиум стало стимулом в разработке ингибиторов, связывающих двойной сайт (рисунок 4.2). (-)-ди(10)-гупиридон проявляет ингибирующую активность 2.4 нМ (Torpedo californica АХЭ), которая является более чем в 200 раз выше по сравнению с мономерным ингибитором АХЭ гуперзином A [27].
Рисунок 4.2 - Молекулярные структуры последовательно кристаллизующихся
ингибиторов АХЭ
4.1.2 Бисубстратные ингибиторы
Другая стратегия использует симметрические и асимметричные двухвалентные лиганды, предназначенные для связывания в кофакторе и сайтах связывания фермента, таким образом, оказывая конкурентное ингибирование. Этот тип лигандов называют бисубстратными ингибиторами. Бисубстратная концепция привела к разработке соединений с мощными лечебными свойствами. Мупироцин (псевдомониевая кислота А) является фемптомолярным (10-15 моль) ингибитором бактериальной изолейцил-тРНК-синтетазы и одним из наиболее широко используемых антибиотиков. Были также обнаружены другие асимметричные бисубстратные ингибиторы для GNC5 - связанной N-ацетилтрансферазы и протеинкиназы.
Юнг и др. применили этот подход, для разработки ингибиторов для фермента
протеин - аргинин - метилтрансферазы (PRMT1). Был получен идентичный ингибитор
PRMT1, который блокирует оба сайта связывания, кофактор S-аденозилметионин
(SAM) и основание (гистон), одновременно (рисунок 4.3). Хотя сайт связывания
асимметричен, симметричный лиганд проявляет благоприятное взаимодействие как с
основанием (гистоном) так и с кофактором сайта связывания [28].
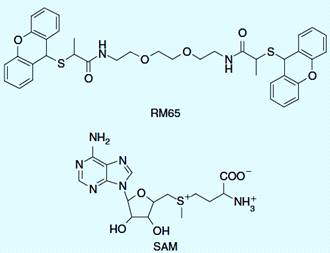
Рисунок 4.3 - Молекулярная структура бисубстратного ингибитора PRMT1 и SAM
.2 Идентичные двойные лекарства, взаимодействующие с двумя подобными
сайтами связывания, расположенными на различных мономерах одной макромолекулы
.2.1 Ингибиторы сиртуина- зависимые деацетилазы гистонов (сиртуины) являются ферментами, которые расщепляют ацетильные группы остатков лизина в гистонах, а также и в других белках. Реверсивное ацетилирование является важным фактором в регулировании деятельности таких белков. Мощные селективные ингибиторы сиртуина - интересные инструменты для исследования биологических функций этих ферментов и могут в будущем использоваться в качестве лекарств для лечения рака. Кристаллическая структура сиртуина подтипа Sirt5 в комплексе с идентичным двойным лекарством сурамином показала, что два белковых мономера связаны одной молекулой сурамина (рисунок 4.4). , который представляет собой в растворе мономер, также был обнаружен в виде димера в растворе после закрепления сурамина, что подтверждается гель - хроматографией. Мономер - мономер взаимодействие в основном неполярное, нет прямых водородных связей между двумя мономерами, таким образом, димерная структура Sirt5, главным образом, стабилизирована самой связанной молекулой сурамина. И в кристаллической структуре, и в растворе, сурамин действует как связывающее звено, приводящее к димеризации Sirt5. Также было отмечено одновременное закрепление одной молекулы сурамина на поверхностях двух мономеров в кристаллической структуре комплекса сурамин - миотоксин II.
Для сиртуинов, однако, это важное открытие может привести к новому классу
ингибиторов, которые не только специфически связывают активный центр, но также
и выполняют функцию связывающего компонента, таким образом, ограничивая подвижность
и доступность фермента. Чтобы получить более мощные и селективные ингибиторы
сиртуина, была изменена структура сурамина. Было обнаружено, что полученные
производные являются мощными ингибиторами сиртуина, с высокой активностью к
подтипу Sirt1. Интересно, что была найдена не только двойная молекула сурамина,
чтобы блокировать Sirt1 (297 nМ),
но также соединение NF154 (рисунок 4.4) - которое напоминает половину структуры
сурамина - мощный ингибитор Sirt1 (525 nM). Исследования показали, что NF154
аналогично взаимодействует с связывающим карманом сиртуина. Однако перекрестное
связывание двух мономеров сиртуина не возможно (рисунок 4.5) [29].
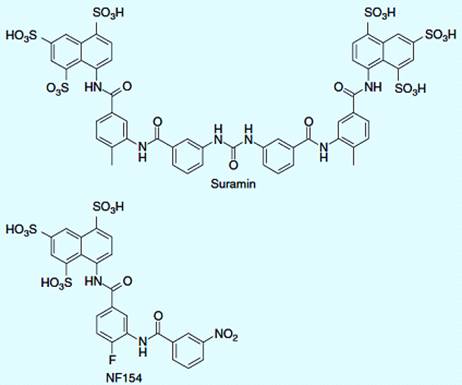
Рисунок 4.4 - Молекулярная структура сурамина и NF 154
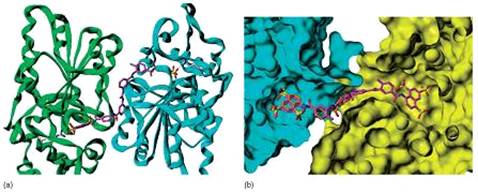
Рисунок 4.5 - (a)
перекрестное связывание двух мономеров Sirt5 (голубой и зеленый) с сурамином (пурпурный).
Оранжевым цветом показаны водородные связи; (b) Связывание сурамина и NF 154
4.2.2 Ингибиторы глутатион S-трансферазы
Глутатион S - трансферазы (GSTs)1 катализируют конъюгацию нуклеофильного трипептида глутатиона (GSH, γ-Glu-Cys-Gly) в структурно различные гидрофобные электрофилы. Среди электрофильных субстратов для GSTs алкилирующие агенты, используемые в химиотерапии рака. GSTs, как известно, повышено продуцируются в злокачественных тканях, предполагая, что они могут играть определенную роль в приобретенной резистентности к противоопухолевым агентам. Поэтому, совместное введение мощных, селективных ингибиторов GST как адъювантов в химиотерапии, появилось в качестве возможной стратегии восстановления лекарственной чувствительности резистентных клеток. Геометрия димера GST, с его двумя идентичными активными центрами на противоположных концах, доступной для растворителя межсубъединичной расщелины, представляет возможность, проектировать симметрические двойные лекарства, которые связывают оба активных центра одновременно.
Имеющаяся кристаллическая структура GST в комплексе с сульфатной
производной антрахинона (рисунок 4.6), использовалась в качестве основы для
разработки новых эффективных ингибиторов. Авторы использовали связанную
молекулу, производную Uniblue А (рисунок 4.7), для создания молекулы двойного
лекарства и чтобы проанализировать свободную энергию связывания [28]. Так как
авторы полагали о близости двух активных центров димера GST и их местоположении
на противоположных концах доступной для растворителя межсубъединичной
расщелина, то предполагалось, что могут быть разработаны двойные ингибиторы,
чтобы одновременно связывать оба активных центра. Соответствующая длина
связывающей цепи, для соединения обоих мономеров основана на исследованиях и
молекулярном моделировании. В то время как мономер производной Uniblue А
показал IC 50 - 5000 nM, двойное лекарство является более чем 100 раз более
активным (IC 50 - 44 nM).
Результаты исследований подтвердили повышенное сродство двойных ингибиторов по
сравнению с мономерными аналогами.
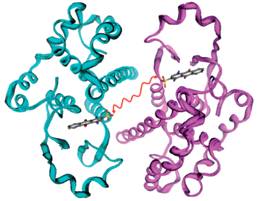
Рисунок 4.6 - Схема комплекса димера GST и сульфатной производной антрахинона. Связывающий
фрагмент между компонентами обозначен красным цветом
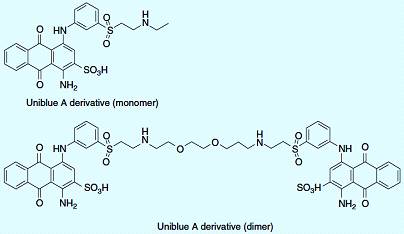
Рисунок 4.7 - Молекулярная структура ингибиторов GST
4.2.3 Лиганды рецепторов, связанных с G-белком
Рецепторы, сопряженные с G-белком (GPCRs) являются мембранными белками,
которые характеризуются общими семью трансмембранными сегментами. GPCRs играют
важную роль во многих биологических процессах. В последние десятилетия, все
больше появилось доказательств, что GPCRs, после активации, димеризуются в
активную форму, а затем оказывают свое биологическое действие. GPCRs находятся
в клеточной мембране или как гомодимеры или как гетеродимеры. Исследования,
показывающие, что гетеродимеризация GPCR может изменить фармакологию рецептора,
вызвали интерес к развитию лекарств, которые селективно нацелены на
гетеродимерные рецепторы. Один из подходов, предназначается для пары GPCRs,
должны быть синтезированы и использованы двойные молекулы, предназначенные для
двух центров связывания рецептора на гомо - или гетеродимере одновременно.
Обоснование использования двойного лекарства к GPCRs выходит из возможности
того, что двухвалентные лиганды способны к связыванию независимого рецептора на
димерном рецепторе, которое приводит к более благоприятным термодинамическим
взаимодействиям, чем связывание двумя одновалентными лигандами. Этот тип
взаимодействия схематично показан на рисунке 4.8.
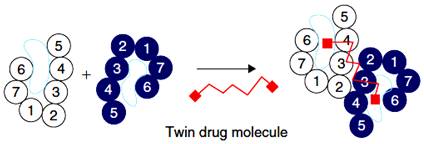
Рисунок 4.8 - Димеризация GPCR и
связывание молекулой двойного лекарства. Двухвалентный лиганд содержит два
обязательных независимых сайта GPCR
Главный прорыв в понимании класса GPCR был достигнут в 2000 году, когда
была открыта кристаллическая структура бычьего родопсина. Структура бычьего
родопсина использовалась различными группами для генерации гомологичных моделей
GPCRs. Эти модели использовались, в руководстве решений конструирования
двухвалентных лигандов, обращенных к двум сайтам связывания GPCR. Portoghese и
др. сообщили о несколько идентичных двойных молекулах с переменной длиной
связывающего фрагмента, предназначенных для исследования фармакодинамики и
особенностей организации опиоидных рецепторов. Подход двойных лекарств, как
было показано, применим и к другим GPCRs. До сих пор нет сведений о двойных
лигандах для рецепторов аденозина, дофамина, гонадотропин-рилизинг гормона,
мелатонина, мускарина, опиоидов, серотонина и вазопрессина [30].
.3 Идентичные и неидентичные двойные лекарства, взаимодействующие с двумя
различными сайтами связывания, расположенными на различных макромолекулах
Лиганды GPCR (гетеродимер). Недавно также было доказано, что многие мономеры различных GPCRs, способны взаимодействовать друг с другом, в результате чего формируются гетеродимеры. Все большее число исследований указывают на роль гетеродимеризации GPCR в модуляции фармакологии рецептора и предполагают, что гетеродимеры могут представлять собой функциональную единицу. Таким образом, создание селективных двойных лигандов может быть перспективной стратегией. Было опубликовано несколько примеров, где селективные гетеродимерные лиганды, оказывали меньше побочных эффектов из-за большей селективности. Portoghese и др. синтезировали серию двойных лигандов димерных опиоидных рецепторов, состоящих из фармакофора антагониста μ - рецептора и фармакофора антагониста δ - рецептора, разделенных прокладками переменной длины. Антиноцицептивная активность двухвалентных лигандов была выше, чем достигнутая путем совместного введения индивидуальных лигандов опиоидных рецепторов.
Будущий анализ закрепления таких лигандов к гетеродимерам и гомодимерам, основанный на структуре моделирования GPCR димеров, обеспечит понимание молекулярных детерминант, необходимых для селективного размещения и/или активации гетеродимеров [31].
Заключение
Лекарства, объединяющие два фармакофора в одной молекуле, описаны в многочисленных областях лекарственной химии. Исторически, их структура подвергалась модификации, и на сегодня рациональный дизайн гомо - и гетеролигандов основан на знании структуры белка, который содержит сайты связывания.
Дизайн двойных лекарств более сложен, чем дизайн соединений с одним видом активности. Наряду с множеством успешных примеров двойных лекарств, могут существовать и некоторые недостатки:
· объединение двух фармакофоров в одной молекуле, может привести к инертному составу. Для успеха подхода необходимо хорошее знание данных о SAR в пределах каждого фармакофора (типы связей, гидрофильные и гидрофобные участки). Также важен выбор связывающего фрагмента (природа, длина).
· гибрид показал ожидаемый фармакологический эффект, но попытка не удалась, из-за не предсказанных фармакодинамических и токсикологических побочных действий.
· должна быть тщательно проанализирована сбалансированность силы лекарственных составляющих. Например, дизайн из агонистов/антагонистов должен принимать во внимание, что лекарства с антагонистической активностью на рецепторы должны быть в значительно меньших концентрациях, чем агонисты (нМ и мкМ соответственно). Подобным образом, при разработке гибридов, состоящих из лиганда рецептора и ингибитора фермента, должны учитываться кинетические свойства рассматриваемого фермента.
Однако, из-за большей терапевтической выгоды, поиск селективных лекарств заменен на дизайн двойных лекарств. В большинстве случаев, одновременное воздействие идентичных двойных лекарств (симметрично связанных участков) связано с увеличением силы воздействия. Димер также проявляет большую селективность, по сравнению с начальным мономером. Объединение в одной молекуле двух неидентичных фармакофоров приводит к новому соединению, обладающему свойствами обоих составляющих.
Таким образом, дизайн двойных лекарств и использование их в качестве ЛС является перспективным и многообещающим подходом в лечении сложных нарушений. На сегодня уже описаны успешные попытки использования двойных лекарств в сердечно-сосудистой области, особенно для лечения гипертонии, в исследованиях ЦНС.
идентичный гомодимер гибридный лекарство
Список использованных источников
1. Contreras J.M., Bourguignon J.J. Identical and non-identical twin drugs. In The Practice of Medicinal Chemistry (Wermuth, C. G., Ed.). Academic Press: London, 2003, pp. 251 - 273.
. Homodimeric tacrine congeners as acetylcholinesterase inhibitors / M.K. Hu [and others] // J. Med. Chem. 2002, 45, 2277 - 2282.
. Neupert-Laves, K., Dobber, M. The crystal structure of a K+ complex of valinomycin. Helv. Chim. Acta. 1975, 58, 432 - 442.
. Pharmacological and behavioral analysis of the effects of some bivalent ligand-based monoamine reuptake inhibitors / A.P. Tamiz [and others] // J. Med. Chem. 2001, 44, 1615 - 1622.
. Structure-activity relationships of methoctramine-related polyamines as muscular nicotinic receptor noncompetitive antagonists. 2. Role of polymethylene chain lengths separating amine functions and of substituents on the terminal nitrogen atoms / M. Rosini [and others] // J. Med. Chem. 2002, 45, 1860 - 1878.
. De Clerq, E. Toward improved anti-HIV chemotherapy: therapeutic strategies for intervention with HIV infections. J. Med. Chem. 1995, 38, 2491 - 2517.
. Design and synthesis of new potent C 2 -symmetric HIV-1 protease inhibitors. Use of the l -mannaric acid as a peptidomimetic scaffold / M. [and others] // J. Med. Chem. 1998, 41, 3782 - 3792.
. Structure, DNA minor groove binding, and base pair specificity of alkyl- and aryl-linked bis(amidinobenzimidazoles) and bis(amidinoindoles) / T.A. Fairley [and others] // J. Med. Chem. 1993, 36, 1746 - 1753.
. Antimalarial, antitrypanosomal, and antileishmanial activities and cytotoxiciy of bis(9-amino-6-chloro-2-methoxyacridines): influence of the linker / S. Girault [and others] // J. Med. Chem. 2000, 43, 2646 - 2654.
. Bridged γ -carbolines and derivatives possessing selective and combined affinity for 5-HT 2 and D 2 receptors / R.E. Mewshaw [and others] // J. Med. Chem. 1993, 36, 1488 - 1495.
. Dibenz[b,e]oxepin derivatives: novel antiallergic agents possessing thromboxane A 2 and histamine H 1 dual antagonizing activity 1/ E. Ohshima [and others] // J. Med. Chem. 1993, 36, 417 - 420.
. Characterization of BIBS 39 and BIBS 222: two new non peptide angiotensine II receptor antagonists / J.C. Zhang [and others] // Eur. J. Pharmacol. 1992, 218, 35 - 41.
. A potent, orally active balanced affinity angiotensin II AT1 antagonist and AT2 binding inhibitor / S.E. de Laszlo [and others] // J. Med. Chem. 1993, 36, 3207 - 3210.