Материал: Современное состояние и перспективы использования двойных лекарств в качестве лекарственных средств
Октапептидный гормон ангиотензин II участвует в сокращении гладких мышц
кровеносных сосудов и в секреции других эндогенных веществ. Т. к. типы
рецепторов ангиотензина II (АТ1 и АТ2) находятся в
различных количествах во многих тканях и органах, двойные антагонисты могут
быть эффективными фармакологическими средствами.
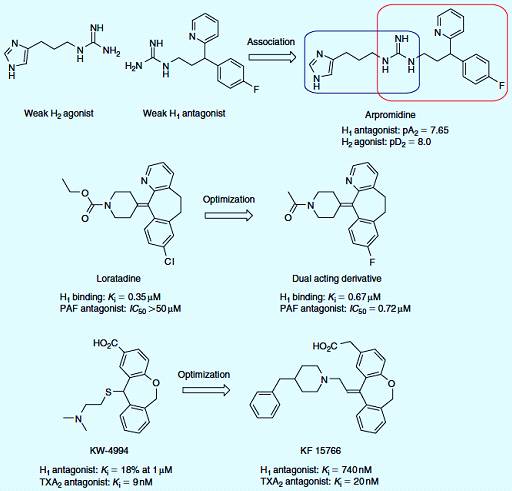
Рисунок 3.1 - Гистаминэргические гибридные лекарства
Начиная с лозартана, селективный антагонист АТ1, и PD 123317, селективный антагонист АТ2, были разработаны двойные неселективные антагонисты АТ1 и АТ2 рецепторов. Структуры лозартана и PD 123317 были объединены с помощью общих структур (синим цветом), и новые подмостки были оптимизированы традиционным исследованием SAR, которое привело к производной ВIВS 222 [12]. Мощный и активный антагонист АТ1 L-159,093, был модифицирован для увеличения сродства к АТ2 рецепторам. После оптимизации, был получен мощный и умеренный антагонист АТ1/АТ2 L-159,689 (рисунок 3.2) [13].
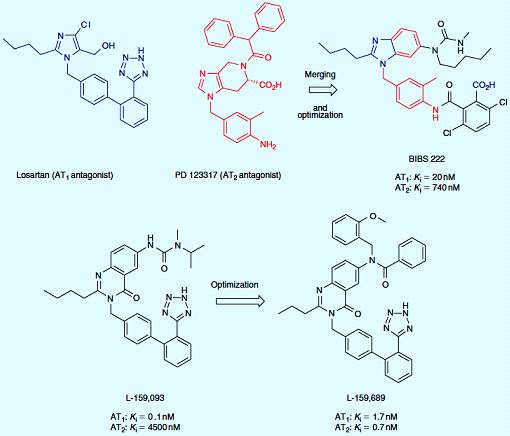
Рисунок 3.2 - Лиганды АТ1/АТ2 рецепторов
Недавние исследования на животных показали, что одновременная блокада АТ1
и АТ2 рецепторов должна быть довольно эффективной в лечении
гипертонии и сердечно-сосудистых заболеваний, таких как сердечная
недостаточность. Бифенилсульфонамид BMS-193884 разработанный как мощный
селективный антагонист ЕТА разделен той же дифенильной структурой
что и ирбесартан, антагонист АТ1. Путем слияния ключевых структурных
элементов этих двух производных, ученые Bristol-Myers Squibb получили гибрид,
который был оптимизирован, для получения производной DARA 7. DARA 7 показал
сбалансированную активность на АТ1 и ЕТА рецепторы и
падение повышенного кровяного давления у крыс (рисунок 3.3) [14].
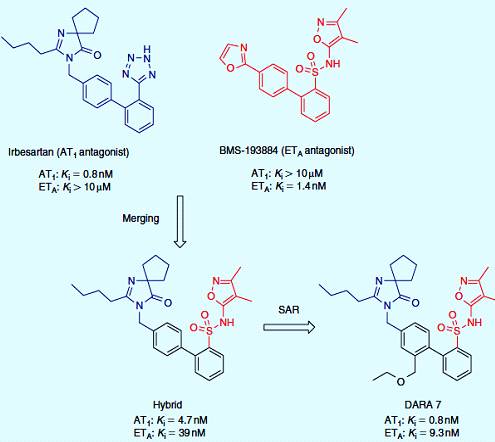
Рисунок 3.3 - Двойные лиганды ангиотензина и ЕТА рецепторов
Лекарства двойного действия также могут быть получены в результате комбинации лигандов, принадлежащих к абсолютно различным фармакофорам. Активация субстанции P (SP) и аденозиновых рецепторов (А1) приводит к тому же эффекту (например, гипотония и обезболивание), что представляет терапевтический интерес к объединению в одном соединении этих свойств. Производная ксантина, антагонист аденозиновых А1 рецепторов, был связан с пентапептидным концевым фрагментом субстанции с получением конъюгата с подобным сродством к А1 и SP рецепторам (рисунок 3.4). Связывание было достигнуто посредством аминокислот [15].
Рецепторы, активируемые пероксисомными пролифератами (PPARs) относятся к ядерным рецепторам, которые включают рецепторы для стероидных, ретиноидых гормонов и гормонов щитовидной железы.
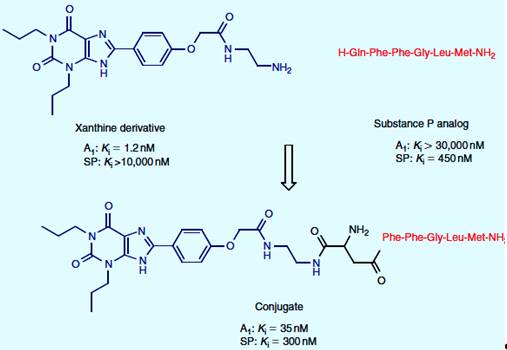
Рисунок 3.4 - Субстанция Р и гибридный лиганд аденозина
Демонстрация того, что PPARα и PPARγ рецепторы, которые являются
посредниками фибратов (гиполипидемическая активность) и глитазона (повышает
чувствительность к инсулину), привела к разработке нового поколения лекарств
двойного действия. Двойные антагонисты PPAR α/γ хорошо подходят для лечения
гипергликемии и профилактики сердечно-сосудистых заболеваний при сахарном
диабете 2 типа. Производные арилоксазола (рисунок 3.5) являются типичным
примером комбинации фенофибриновой кислоты и производной тиазолидинона
Фенофибриновая кислота, антагонист PPARα, была объединена с селективным PPARγ
производным
тиазолидинона; гибридное производное получено замещением фенильной группы, для
увеличения силы. Дальнейшие исследования SAR привели к разработке более мощного
антагониста двойного действия PPARα и PPARγ, такого как мураглитазар. С другой
стороны, оптимизация производных α - замещенной фенилпропановой кислоты
приводит к получению двойных агонистов PPAR α/PPAR δ
[16].
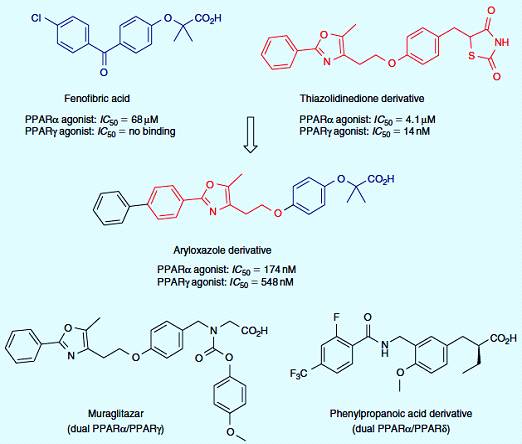
Рисунок 3.5 - Двойные агонисты PPAR
3.2 Гибридные молекулы как ингибиторы ферментов
Как и рецепторы, ферментативные системы можно разделить на классы ферментов, и каждый тип ферментов представлен несколькими изоформами. Таким образом, в фармакологических и терапевтических целях, это может представлять интерес, для объединения в одной молекуле структурных ингибиторов двух различных изоферментов, двух ферментов, принадлежащих к одному классу или двух ферментов, ингибиторы которых обладают сходными фармакофорными свойствами.
Ингибиторы циклооксигеназы (СОХ-2) и 5-липооксигеназы (5-LO), ферменты,
участвующие в биосинтезе простагландинов и лейкотриенов, изучаются как
нестероидные противовоспалительные средства с повышенным уровнем безопасности.
Соединения двойного действия с двойным ингибирующим действием, используются для
лечения пациентов, страдающих от артрита и других заболеваний. Тиазолон CI-1004
(рисунок 3.6) был идентифицирован как аналогичный двойной ингибитор COX/5-LO.
По сравнению с соединением KME-4, этот препарат двойного действия
неульцерогенный, водорастворимый и при пероральном введении проявляет
противовоспалительные свойства. В последнее время, было достигнуто соединение
фармакофоров селективных ингибиторов СОХ-2 и 5-LO. Новое сопряженное производное
показало мощное ингибирование COX-2/5-LO и высокую селективность к СОХ-2 [17].
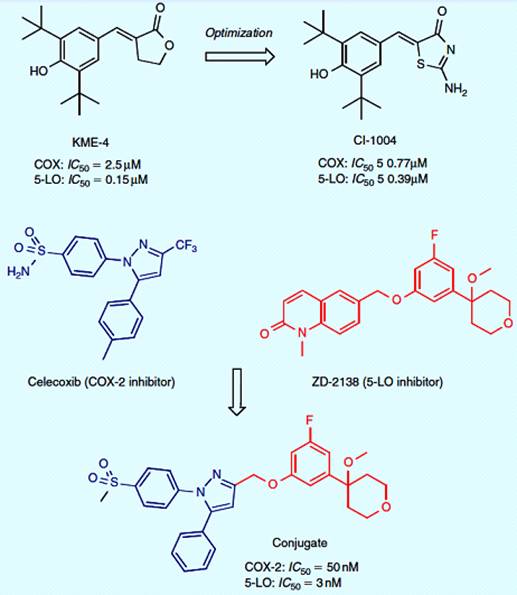
Рисунок 3.6 - Двойной ингибитор СОХ/5-LO
Инактивация эндогенного опиоидного пептида энкефалина является одной из
физиологических функций нейтральных эндопептидаз (НЭП). Было предположено, что
одновременное ингибирование ангиотензин превращающего фермента (АПФ) и НЭП
может быть выгодным для лечения застойной сердечной недостаточности или
гипертензии. Тиорфан, известный ингибитор НЭП, имеет двойные ингибирующие
АПФ/НЭП свойства, но как ингибитор НЭП он в сотни раз мощнее, чем как ингибитор
АПФ (рисунок 3.7). Жесткий бензазепинон был разработан как PheLeu миметик и
показал мощное двойное ингибирование НЭП/АПФ. Исследования в области изучения
ингибиторов двойного ряда привели к проектированию бициклического тиазепинона
омапатрилата. Это соединение показало равносильное двойное ингибирование
НЭП/АПФ и продемонстрировало хорошие результаты в снижении давления у животных.
Омапатрилат был разработан для лечения гипертонии и застойной сердечной
недостаточности [18].
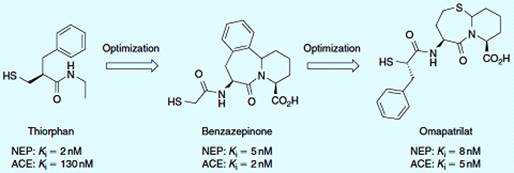
Рисунок 3.7 - Двойные ингибиторы НЭП/АПФ
Нуклеозидные ингибиторы обратной транскриптазы (NRTIs), такие как
зидовудин или зальцитабин показали высокую эффективность, когда они
используются в сочетании с ингибиторами PR ВИЧ. Из-за увеличения
неблагоприятных побочных эффектов, исследование было сосредоточено на
ненуклеозидных ингибиторах обратной транскриптазы (NNRTIs),
неконкурентоспособных ингибиторах сайта связывания. Комбинированная терапия
NRTI-NNRTI проявит синергическую активность и имеет большую эффективность.
Таким образом, соединения, содержащие как нуклеозидные, так и ненуклеозидные
ингибиторы (рисунок 3.8), были разработаны и показали микромолярную анти-ВИЧ
активность. Производная гетеродимер не показала синергетический эффект,
предполагается, что отдельные фармакофоры связываются не одновременно [19].
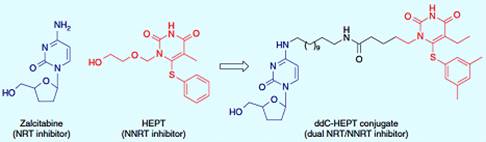
Рисунок 3.8 - Гетеродимер ингибитор ВИЧ
Назначение ингибиторов ацетилхолинэстеразы (АХЭ) представляет
перспективный подход для лечения болезни Альцгеймера, так как была
продемонстрирована связь между этим ферментом и образование амилоида. Мощный и
селективный ингибитор АХЭ был разработан в результате слияния двух различных
ингибиторов АХЭ гуперзина A и такрина (рисунок 3.9). Гибридная производная
показала улучшенную ингибирующую активность с умеренной селективностью. Лиганды
двойного взаимодействия с галантамином были получены при помощи различных
метиленовых связей из фрагмента фталимида и производной галантамина, ингибитора
активного центра. Биохимическое исследование кристаллографической структуры АХЭ
позволило четко идентифицировать два связывающих участка на ферменте: активный
центр, расположенный у основания глубокого и узкого ущелья, и периферический
сайт, расположенный при открытии ущелья. Комбинация фрагмента фталимида,
который, как известно, взаимодействует с периферическим сайтом, с производной
галантамина (ингибитор активного центра), привела к соединению, которое
связывается и с активным и с периферическим центрами [20].
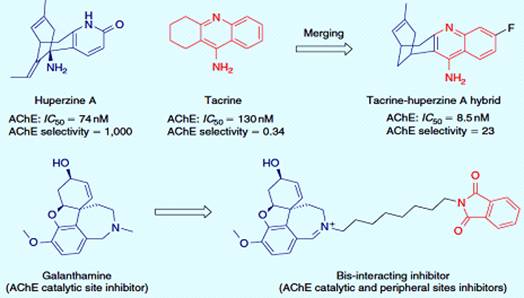
Рисунок 3.9 - Гибридный ингибитор АХЭ
В дополнение к дефициту холинергической нейротрансмиссии, наблюдаемом в
н. э. и коррелированном с когнитивными нарушениями функций, пациенты в наше
время также страдают от депрессии и беспокойства. Для лечения этих симптомов
используются ингибиторы транспортеров серотонина (SERT). Создание двойных
ингибиторов (АХЭ и SERT) было достигнуто, для получения лучшего
терапевтического эффекта. После гибридизации ривастигмина, ингибитора АХЭ, и
флуоксетина, мощного ингибитора SERT, был создан новый двойной ингибитор АХЭ и
SERT (рисунок 3.10). После исследований SAR, было получено соединение ((R) -
аналог) с мощным ингибирующим действием на оба фермента [21].
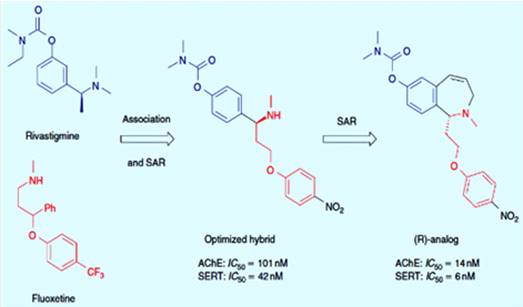
Рисунок 3.10 - Двойной ингибитор АХЭ/SERT
3.3 Гибридные молекулы, действующие на один рецептор и фермент
Гибридные молекулы, действующие одновременно на рецептор и на фермент,
могут привести к мощным синергетическим эффектам. Рассмотрим на примере
производных взаимодействующих с TXA2, мощным индуктором агрегации
тромбоцитов и сокращения гладких мышц кровеносных сосудов. Ингибирование TXA2
синтазы (TxS) и селективная блокада TXA2 рецептора (TxR)
рассматривается в качестве альтернативной терапевтической стратегии по
предотвращению тромбического действия TXA2. Таким образом, ингибитор
TxS и антагонист TxR были объединены в одну молекулу, такие как самиксогрел,
ингибитор синтазы, и далтробан, антагонист TXA2 рецептора (рисунок
3.11). Поскольку самиксогрел показал умеренные плазменные уровни после
перорального приема (низкая растворимость в водном растворе), то он был
оптимизирован в производную гуанидина тербогрел, который проявляет более мощные
антитромбические эффекты in vivo [22].
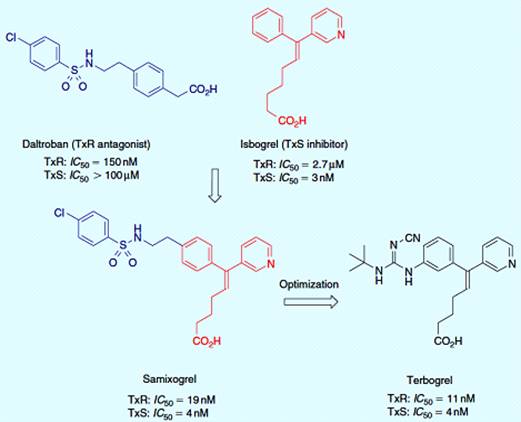
Рисунок 3.11 - Гибридное лекарство ТХА2
Основываясь на физиологической гипотезе, обе мишени могут принадлежать к
различным системам. Дигидропиридины, такие как нифедипин, как известно,
являются антагонистами кальциевых рецепторов кальция (Ca2+). Эти
препараты обычно используются для лечения пациентов с сердечно-сосудистыми
заболеваниями (гипертония, инфаркт миокарда). Сочетание антагониста Ca2+ и
ингибитора TxS может повысить терапевтическую эффективность для конкретных
патологий, где, и повышен синтез TXA2 и наблюдается клеточная
перегрузка кальцием. Имидазольный фрагмент дазоксибена, ингибитора TxS, был
слит с основной структурой нифедипина, что привело к образованию FEC 24265
(рисунок 3.12). Гибридная производная показала более благоприятный
фармакологический эффект in vivo. Фактор активации тромбоцитов (PAF)
участвует в процессе воспаления и связан с патологиями, такими как ишемия,
тромбоз и астма. Объединение антагониста PAF и ингибитора TxS обеспечило бы
благоприятное терапевтическое лечение. Ридогрел, мощный ингибитор TxS, был
непосредственно связан с антагонистом PAF Е-6123. Гетеродимерный лиганд показал
двойное равносильное сродство и ингибирующую способность к PAF и TxS,
выраженную активность после перорального приема [23].
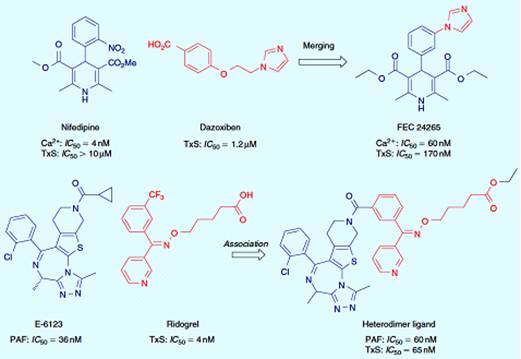
Рисунок 3.12 - Гибридные лекарства с синергическим эффектом
Новые антигипертензивные средства, обладающие β
- блокирующими и
ингибирующими ангиотензин превращающий фермент (АПФ) свойствами представляют
благоприятный подход для лечения повышенного кровяного давления. Гибридизация β
- блокатора пиндолола и
ингибитора АПФ энелаприла привела к производному BW-A575C, который обладает
двумя действиями (рисунок 3.13). Эти двойные лекарства, β
- блокатор/ингибитор
АПФ, терапевтически выгодны при лечении гипертонии и застойной сердечной
недостаточности [24].
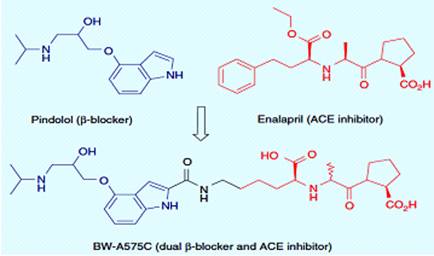
Рисунок 3.13 - Двойной ингибитор АПФ/β - блокатор
3.4 Другие примеры лекарств двойного действия
Комбинированное лечение необходимо при длительном лечении гипертонической
болезни. β - блокирующие и мочегонные свойства в одной молекуле
представляют большой интерес при гипертонии напряжения. Описано мало попыток
синтезировать гибридные молекулы, объединяя структуры антагонистов β-АР и мочегонных средств (рисунок
3.14). Гибридный сульфонамид был получен, путем связывания β
- блокатор производного
пропранолола с 2-хлорбензол сульфамидным фрагментом мефрузида [25].
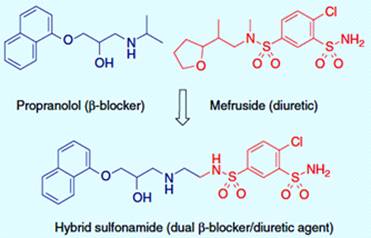
Рисунок 3.14 - Гипотензивные гибридные лекарства
Антибактериальные лекарства двойного действия были разработаны, связывая
хинолоны (рисунок 3.15), такие как ципрофлоксатин, в цефотаксим. Гибридный
лиганд был оптимизирован и продемонстрировал мощную активность против широкого
спектра грамположительных и грамотрицательных бактерий.
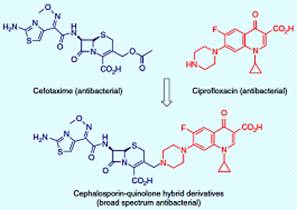
Рисунок 3.15 - Антибактериальные гибридные лекарства
В результате поиска новых эффективных антидепрессантов, были разработаны
лекарства двойного действия с селективным ингибированием серотониновых
рецепторов (SSR) и антагонизмом 5-HT1А рецептора (рисунок 3.16).
Комбинация ингибитора SSR дулоксетина и производных арилпиперазина, которые,
как известно, имеют высокое сродство к 5-HT1А рецепторам, привела к
образованию гибридов бензотиофенпиперазина, класса потенциальных
антидепрессантов с двойным механизмом действия [26].
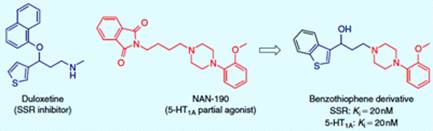
Рисунок 3.16 - Гибридный антидепрессант
4. Способ закрепления идентичных и неидентичных двойных лекарств к
ассиметричному центру связывания
.1 Идентичные и неидентичные двойные лекарства, взаимодействующие с двумя
смежными центрами связывания, расположенными на одной макромолекуле
.1.1 Ингибиторы ацетилхолинэстеразы
Трехмерная структура АХЭ из электрического органа ската (Torpedo californica) показала, что активный центр расположен у основания глубокого и узкого ущелья (20 Å), выровненный кольцами нескольких ароматических аминокислот. На “анионном подместе” активного центра, моделированием было предположено, что четвертичная аминогруппа ацетилхолина связывается с индольной боковой цепью сохраненного остатка, Trp84, как было продемонстрировано в дальнейшем для нескольких комплексов с АХЭ. Комплексы АХЭ с дичетвертичными лигандами (дэкаментоний и BW284C51) привели к назначению Trp279 как основного элемента второго сайта связывания, в верхней части ущелья активного центра, названное “периферическим сайтом связывания, в 14 Å от активного центра.
Эти структурные назначения были подтверждены большим числом биохимических исследований, включая сайт направленный мутагенез, которые подтвердил важность остатков ароматических аминокислот в АХЭ. Чтобы улучшить силу и селективность препарата, была применена стратегия двойных лекарств к созданию ингибитора двойного действия на центр АХЭ. Несколько идентичных димеров такрина, например ди(5)-такрин и ди(7)-такрин, были синтезированы и оценены после того, как вычислительные исследования показали слабое сродство такрина к остаткам периферического сайта связывания (Trp279 и Tyr70) в сочетании с высоким сродством такрина к анионному каталитическому сайту (Trp84 и Phe330).