Материал: Потапов В.В. Решение задач биоинформатики при помощи веб - и интернет-сервисов

ATOM |
14 |
O |
HIS A |
45 |
19.550 |
1.921 |
20.345 |
1.00 |
10.73 |
O |
ATOM |
15 |
CB |
HIS A |
45 |
20.548 |
4.597 |
20.226 |
1.00 |
6.82 |
C |
ATOM |
16 |
CG |
HIS A |
45 |
21.635 |
4.402 |
21.239 |
1.00 |
5.48 |
C |
ATOM |
17 |
ND1 HIS A |
45 |
22.138 |
5.425 |
21.999 |
1.00 |
1.29 |
N |
|
ATOM |
18 |
CD2 HIS A |
45 |
22.253 |
3.277 |
21.664 |
1.00 |
7.20 |
C |
|
ATOM |
19 |
CE1 HIS A |
45 |
23.085 |
4.962 |
22.805 |
1.00 |
4.74 |
C |
|
ATOM |
20 |
NE2 |
HIS A |
45 |
23.134 |
3.645 |
22.643 |
1.00 |
4.10 |
N |
ATOM |
21 |
N |
MET A |
46 |
18.357 |
2.463 |
22.204 |
1.00 |
9.56 |
N |
ATOM |
22 |
CA AMET A |
46 |
18.138 |
1.082 |
22.631 |
0.50 |
12.15 |
C |
|
ATOM |
23 |
CA BMET A |
46 |
18.139 |
1.096 |
22.655 |
0.50 |
12.59 |
C |
|
ATOM |
24 |
C |
MET A |
46 |
16.721 |
0.700 |
22.236 |
1.00 |
13.12 |
C |
ATOM |
25 |
O |
MET A |
46 |
15.855 |
1.558 |
22.093 |
1.00 |
14.29 |
O |
Структура представления данных в модели достаточно проста - первый столбик обозначает, что речь идет об атоме, далее следует его порядковый номер, далее номенклатура атомов в аминокислоте, сама аминокислота (GLY, SER, HIS...), имя цепи, порядковый номер аминокислоты в цепи, пространственные координаты этого атома и другая информация, однозначно характеризующая его положение и свойства, подробнее можно просмотреть в Интернете по адресу http://www.wwpdb.org/documentation/format32/v3.2.html
Задание: Найдите, как в тексте формата PDB кодируется вторая цепь аминокислот (мы видели, что их две в программе UCSF Chimera)
Кроме загрузки и сохранения собственно трехмерной модели молекулы, можно также сохранить её пространственное положение и раскраску. Это часто требуется для демонстрационных целей или при подготовке научных статей. Для сохранения правильно повернутой и любовно раскрашенной модели нужно выбрать File/Save session (Файл/сохранить сессию), для загрузки, соответственно File/Restore session (Файл — восстановить сессию)
3.2Редактор JalView и FASTA - формат
Редактор JalView (Java ALigned VIEWer) предназначен для ручного и полуавтоматического парного и множественного выравнивания последовательностей. Создан он в университете Данди, Шотландия. Как и для UCSF Chimera, для полного раскрытия функциональности программы нужно подключение к сети Интернет.
Скачать инсталляционные файлы этой программы можно по адресу http://www.jalview.org/download.html
16
При написании пособия использовался JalView версии 2.6.1, если у вас будет более свежая версия, возможны изменения в интерфейсе. При первом старте программы она загружает демонстрационные данные, чтоб это отключить, снимите галочку в меню Tools/Preferences/Open file. Если доступ к Интернету осуществляется через проксисервер, возможно понадобится установить параметры прокси в
Tools/Preferences/Connectons
Установим и запустим программу. Для того, чтоб ознакомиться с ее работой, нужно загрузить в нее какую-то последовательность. Какую? Ну, например ту самую, на основе которой была построена пространственная модель чуть раньше, в UCSF Chimera.
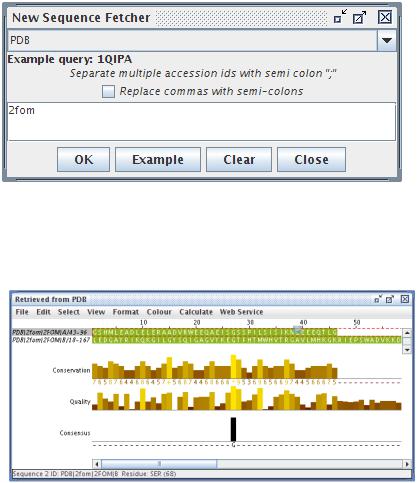
Выбираем из меню File/Fetch Sequence(s) (Файл — получить последовательность(и)), переключаем в открывшемся окне на базу PDB и вводим уже знакомый нам идентификатор последовательности 2fom.
Рисунок 7: JalView - Загрузка последовательности из базы данных по идентификатору
После клика на OK программа загрузит из базы данных пространственную модель, выберет из нее последовательности (мы видели, как они хранятся в модели) и откроет окно
Рисунок 8: JalView - окно для работы с последовательностями
В этом окне мы можем наблюдать обе цепи, верхняя линейка показывает позиции каждой аминокислоты в последовательности. Т.к. JalView используется в том числе для оценки множественного выравнивания, внизу приведена оценка этого выравнивания для двух наших последовательностей. Очевидно, что она в нашем случае не имеет никакого смысла и ее можно выключить, убрав галочку в View/Show annotation (просмотр/показывать аннотацию)
Теперь сохраним цепи.
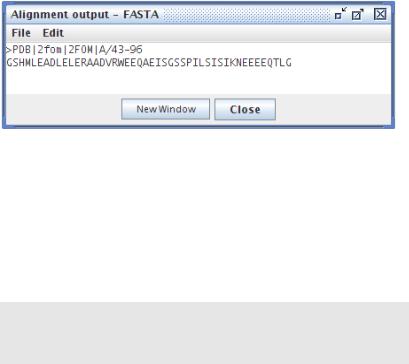
Выделим верхнюю цепь, щелкнем правой кнопкой мыши Selection/Out to text box/FASTA (выделение — вывод в текстовое поле/формат FASTA)
Получим на экране самый распространенный, пожалуй, формат представления биологических последовательностей — FASTA-формат.
Рисунок 9: JalView - вывод последовательности в FASTA-формат
В первой его строчке, после знака > идет название последовательности, здесь чаще всего пишется база, из которой получена последовательность и код этой последовательности в базе. Потом же идет сама биологическая последовательность, она может быть аминокислотной или генетической.
Сохранить данные на диск можно традиционным образом, через
File/Save.
Задание: Сохраните вторую последовательность в другой FASTAфайл. Найдите как кодируются в формате FASTA несколько биологических последовательностей, сохраните две наших последовательности в один FASTA-файл
18
3.3Контрольные вопросы
1.Для чего применяется программа UCSF Chimera?
2.Что объединяет форматы файлов пространственных структур (PDB) и биологических последовательностей (FASTA)?
3.Зачем нужен заголовок в FASTA-файле?
4.Для чего может понадобиться в UCSF Chimera раскраска различных участков модели разным цветом?
5.Для чего применяется программа JalView?
6.Как хранится информация о координатах атомов в пространственной модели?
7.Как в одном FASTA-файле хранятся несколько последовательностей?
4Интернет- и веб-сервисы: основные понятия, проблемы и перспективы
4.1Принципы работы интернет- и веб-сервисов
Даже если вы никогда не слышали слов «интернет-сервис» и «вебсервис», вы, скорее всего, ими пользовались. Интернет - сервис, это установленная на вашем компьютере программа или ее часть, которая использует для своих нужд связь с интернетом. Чаще всего, часть программы, установленная на компьютере пользователя, называется клиентом, а та часть, что предоставляет данные где-то в Интернете - сервером. Клиент запрашивает какую-то информацию, а сервер её предоставляет. В UCSF Chimera и JalView мы уже пользовались их интернет-сервисами для загрузки данных в программу из интернет-баз. Вы скорее всего знакомы с такими интернет-сервисами, как программы мгновенной передачи сообщений ICQ или Skype.
Интернет первоначально был создан для показа ссылающихся друг на друга страниц через браузер (Internet Explorer, Mozilla Firefox и др.) но, с усложнением технологий его использования, стало возможно выполнять прямо в окне браузера довольно сложные приложения.
19
Такие приложения называются веб-сервисами. Самые известные вебсервисы это, конечно, поисковики, например google или yandex, также известны почтовые сервисы типа gmail или mail.ru. Общий принцип различия интернет- и вебсервисов прост: если вы работаете с чем-то интернетным через браузер, заходя на странички www — это вебсервис, а если вы работаете в программе, а она уже в свою очередь както общается с Интернетом — это интернет-сервис.
Зачем мы останавливаемся на этом вопросе так подробно? Затем, что сегодняшние средства для решения задач биоинформатики, которые начинались с маленьких разрозненных программ, решающих какую-то одну задачу, развиваются по двум этим направлениям. Либо отдельные программы, совершенствуясь в процессе «эволюции» «обрастают» всяческими средствами для работ с интернет-базами и сервисами (как те же UCSF Chimera, JalView, можно добавить сюда и новосибирскую разработку - Ugene), либо разрозненные утилиты собираются где-то на интернет-сервере и делается веб-интерфейс для работы с ними через браузер. Так сделаны, например, российско — белорусский сервис http://bri-shur.com или французкий http://mobyle.pasteur.fr
Оба подхода имеют свои плюсы и минусы. Интернет-сервисы требуют установки на компьютер и постоянного обновления программ, зато у них гораздо более развиты средства визуализации. Веб-сервисы доступны с любого компьютера, где есть браузер, но полноценная работа, например с пространственными моделями в графическом виде
вбиологических веб-сервисах пока не реализована.
4.2Выравнивание последовательностей в MAFFT
Одна из наиболее часто встречающихся в биоинформатике задач — задача выравнивания биологических последовательностей.
Выравнивание последовательностей — взаимное размещение последовательностей ДНК, РНК, и белков для того, чтобы увидеть места схожести, которые могут быть следствием функциональных, структурных или эволюционных взаимосвязей между этими последовательностями.
Различают два вида выравнивания: парное, когда выравниваются две последовательности и множественное, когда этих последовательностей несколько.
20