Материал: Инфаркт миокарда
. Острое уменьшение сердечного выброса вследствие прекращения сократительной деятельности некротизирующегося участка миокарда является одной из причин развития, как преходящей артериальной гипотензии, так и необратимого кардиогенного шока. Этим же объясняют и развитие при инфаркте миокарда сердечной астмы, отёка лёгких, а также поражений внутренних органов: головного мозга (нарушения мозгового кровообращения), желудочно-кишечного тракта (эрозивный гастрит, панкреатит), печени, почек и другие. Вместе с тем в поражении указанных органов играют роль повышенное выделение катехоламинов, вызывающих спазм сосудов, а также висцеро-висцеральные рефлексы (например, парез желудка и кишечника) и, возможно, токсическое влияние продуктов распада некротического очага.
. Развитие резорбционно-некротического синдрома и асептического воспаления. Из некротизированных клеток миокарда высвобождаются ферменты и токсические продукты, которые вызывают повышение температуры, нейтрофильный лейкоцитоз с палочкоядерным сдвигом, а в дальнейшем ускорение СОЭ.
. Перераспределение электролитов в области, прилежащей к очагу некроза (периинфарктная зона). Нарушение баланса внутри и внеклеточного K+ и Na+ ведёт к возникновению электрической нестабильности миокарда в периинфарктной зоне, что создаёт предпосылки к развитию нарушений ритма сердца: желудочковой экстрасистолии, желудочковой тахикардии, фибрилляции желудочков и другим видам аритмий сердца.
. Распространение некроза или периинфарктной зоны на проводниковую систему сердца формирует преходящие или стабильные нарушения проводимости, которые при инфаркте миокарда могут локализоваться на разных уровнях проводниковой системы. Ишемия или некроз синусового узла служат предпосылкой к возникновению ряда аритмий (например, мерцания или трепетания предсердий и другие) или остановки сердца.
Кроме перечисленных патогенетических факторов, определяющих основные, наиболее общие клинический, проявления крупноочагового инфаркта миокарда, в ряде случаев действуют и другие, определяющие частные клинический, симптомы. Повышенное конечное диастолическое давление при обширном и глубоком очаге некроза создаёт предпосылки к развитию острой или подострой аневризмы сердца, а также к разрыву миокарда. Наличие мелких очагов дистрофии или некроза миокарда на отдалении от основного, крупного очага является возможной причиной возникновения при инфаркте миокарда мерцательной аритмии, которая может быть также следствием острой перегрузки и перенапряжения левого предсердия из-за недостаточности левого желудочка. Распространение некроза на сосочковую мышцу ведёт к тяжёлой застойной левожелудочковой недостаточности, так как поражение сосочковой мышцы служит причиной недостаточности митрального клапана за счёт пролабирования его створки в полость левого предсердия во время систолы желудочков
Правожелудочковая недостаточность, которая в остром периоде инфаркта миокарда наблюдается редко, может быть связана с несколькими причинами:
) инфаркт правого желудочка или распространение инфаркта левого желудочка на правый;
) разрыв межжелудочковой перегородки;
) аневризма межжелудочковой перегородки с выпячиванием её в полость правого желудочка (синдром Бернгейма);
5.Патологическая
анатомия и физиология инфаркта
Инфаркт является не мгновенным событием, а продолжительным процессом усугубляющейся ишемии, которая, в конце концов, выливается в гибель клеток. Миокард, кровоснабжаемый окклюзированным сосудом, погибает быстро. Прилежащие ткани, частично кровоснабжаемые соседними артериями, некротизируются не сразу. С течением времени, однако, ишемия соседних клеток прогрессирует, поскольку потребность в кислороде остается высокой, а его доставка сниженной. Таким образом, зона некроза может постепенно расширяться. Масса миокарда, в конце концов, подвергающегося некрозу, таким образом, зависит от:
) размера зоны, кровоснабжаемой окклюзированным сосудом;
) ее потребности в кислороде;
) адекватности коллатерального кровотока, осуществляемого соседними неокклюзированными артериями;
) выраженности ответа ткани на ишемию.
Эффективность терапевтического вмешательства и возможные осложнения зависят от особенностей патофизиологических процессов, развивающихся во время инфаркта миокарда. В целом, эти процессы можно разделить на две стадии: ранние изменения, развивающиеся непосредственно во время инфаркта, и поздние изменения, происходящие в восстановительном периоде.
РАННИЕ ИЗМЕНЕНИЯ
Ранние изменения включают гистологическую эволюцию инфаркта и функциональные изменения вследствие влияния недостатка кислорода на сократимость. Кульминацией этих изменений является коагуляционный некроз миокарда, максимально выраженный на 2-4 сутки (рис.1).
- Цитологические изменения
Падение уровня кислорода в миокарде при острой окклюзии сосуда приводит к быстрому переходу от аэробного обмена к анаэробному, гликолитическому. Поскольку окисление жиров и продуктов гликолиза в митохондриях становится невозможным, синтез макроэргических фосфатов резко снижается, а анаэробный гликолиз ведет к накоплению лактата. Снижение рН обусловливает уменьшение податливости и сократимости миокарда уже через 2 минуты после окклюзии сосуда. При отсутствии лечения через 20 минут развивается необратимое повреждение клеток, проявляющееся набуханием митохондрий, краевым расположением ядерного хроматина, дефектами мембраны и истощением запасов гликогена.
В течение нескольких минут
содержание АТФ в клетке падает, так как его продукция в результате гликолиза
далеко не обеспечивает потребности в нем. Недостаток АТФ подавляет активность
трансмембранной Na+/K+-АТФазы, в результате чего повышаются уровни внутриклеточного Na+
и внеклеточного К+. Повышение Na+ усугубляет отек клетки.
Дефекты мембраны и повышение внеклеточного К+ приводят к изменению
трансмембранного потенциала, создавая условия для возникновения фатальных
аритмий.
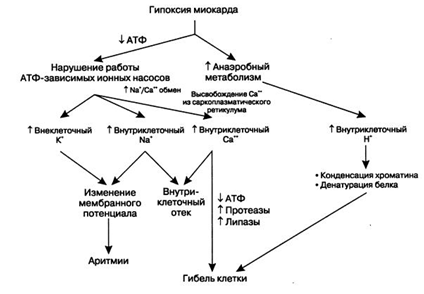
Рис. 1. Механизмы гибели
клеток при инфаркте миокарда. При
острой ишемии прекращается аэробный гликолиз и быстро падает клеточный запас
АТФ. Это влечет за собой внутриклеточный ацидоз и нарушение АТФ-зависймых
процессов, что в свою очередь вызывает накопление в клетке кальция, отек и
гибель клетки.
Во время ишемии внутри клетки в результате ряда причин накапливается Са2+; к этим причинам относятся: 1) активация Na+/ Са2+-ионообменного насоса вследствие повышения концентрации Na+ внутри клетки, 2) просачивание Са2+ из саркоплазматического ретикулума в цитозоль и 3) изменения в работе потенциалзависимых Са2+-каналов и Са2+-АТФазы. Вследствие прогрессирующей деструкции клеточной мембраны Са2+, поступающий из экстрацеллюлярного пространства, не может быть удален нормальными энергозависимыми механизмами, что отражает переход от обратимого к необратимому повреждению клеток. Высокая внутриклеточная концентрация ионов Са2+ считается единым конечным механизмом, приводящим к деструкции клеток миокарда липазами и протеазами. Тяжелые дефекты клеточных мембран возникают вследствие недостатка макроэргических фосфатов, потери эндогенных антиоксидантов и продукции свободных радикалов нейтрофилами. Протеолитические ферменты, просачивающиеся из некротизированных миоцитов, повреждают соседний миокард; некоторые макромолекулы, попадающие в кровоток, являются диагностическими маркерами острого инфаркта.
В течение 4-12 часов, по мере роста проницаемости сосудов под действием медиаторов воспаления и повышения онкотического давления в интерстиции (вследствие просачивания туда внутриклеточных белков), развивается отек миокарда. Наиболее ранним гистологическим изменением, характерными для необратимого повреждения, является волнистость миофибрилл, которая возникает в результате интерстициального отека и "растягивания" в разные стороны кардиомиоцитов под действием сокращений окружающего миокарда. По краю инфарктной зоны часто видны пояски сокращения: сократившиеся и консолидированные саркомеры выглядят как яркие эозинофильные полоски.
Приблизительно через 4 часа начинается воспалительный ответ на повреждение, включающий инфильтрацию нейтрофилами, продуцирующими
токсичные свободные радикалы, усугубляющими повреждение ткани. Через 18-24 часа при световой микроскопии видны такие признаки коагуляционного некроза, как кариопикноз и слабо выраженная эозинофилия цитоплазмы. Эти ранние изменения представлены в таблице 1.
Макроскопические изменения
На макропрепарате не удается увидеть никаких изменений в течение 18-24 часов после окклюзии артерии, хотя некоторые методики окраски (в частности, тетразолием) позволяют идентифицировать зоны инфаркта и ранее. Наиболее часто ишемия и инфаркт начинаются с субэндокардиальных слоев и затем распространяются в стороны и по направлению к эпикарду.
ПОЗДНИЕ ИЗМЕНЕНИЯ
Поздние патоморфологические изменения, характерные для дальнейшего течения острого инфаркта (табл. 1), включают: 1) очищение миокарда от некротических масс макрофагами и 2) отложение коллагена и формирование рубцовой ткани.
Необратимо поврежденные
кардиомиоциты не регенерируют, они резорбируются и замещаются рубцовой тканью.
Макрофаги проникают в воспаленный миокард вскоре после инфильтрации его
нейтрофилами и удаляют некротическую ткань. Этот период резорбции ткани, когда
наряду с мертвыми кардиомиоцитами разрушаются и удаляются элементы
соединительной ткани, называется желтым размягчением. Активный фагоцитоз в
сочетании с истончением и растяжением зоны инфаркта на этой стадии приводит к
структурной слабости стенки желудочка и повышению риска ее разрыва.
Впоследствии развивается фиброз и через 7 недель после инфаркта формирование
рубца заканчивается.
Таблица 1. Динамика патологических изменений при трансмуральном инфаркте


ФУНКЦИОНАЛЬНЫЕ ИЗМЕНЕНИЯ:
1)НАРУШЕНИЕ СОКРАТИМОСТИ
Инфаркт миокарда быстро приводит к снижению сократительной функции желудочка и уменьшению сердечного выброса. Далее сердечный выброс еще больше снижается из-за потери синхронности сокращения миоцитов: если инфаркт приводит к снижению сократимости, зона инфаркта характеризуется как "гипокинетичная", вообще не сокращающиеся сегменты называются "акинетичными", а "дискинетичные" сегменты в систолу выбухают наружу.
)"ОГЛУШЕННЫЙ" И "СПЯЩИЙ" МИОКАРД
Ранее считалось, что ишемическое повреждение миокарда приводит либо к необратимому некрозу миокарда, либо к быстрому полному восстановлению функции кардиомиоцитов. Теперь стало известно, что ишемические атаки иногда могут приводить к длительной сократительной дисфункции без некроза кардиомиоцитов, и впоследствии их функция может полностью восстанавливаться.
Когда после периода тяжелой ишемии (но не некроза), даже после восстановления нормального коронарного кровотока, длительно сохраняется систолическая дисфункция, говорят об "оглушенном" (станнирующем) миокарде. В этом случае функциональные, биохимические и ультраструктурные ишемические изменения обратимы, и сократимость постепенно восстанавливается. Механизм этого отсроченного восстановления функции пока не известен, но возможно он связан с перегрузкой кардиомиоцитов кальцием, накоплением свободных радикалов или электромеханической диссоциацией вследствие транзиторной дисфункции саркоплазматического ретикулума. В целом выраженность "оглушенности" пропорциональна степени предшествующей ишемии. Таким образом, "оглушенный" миокард, видимо, является патофизиологическим ответом на ишемическое повреждение, недостаточное для развития необратимого некроза.
Гибернирующий ("спящий") миокард отличается от "оглушенного". Этим термином обозначают хроническую сократительную дисфункцию миокарда в условиях стойкого снижения коронарного кровотока, обычно при многососудистом поражении. В этой ситуации отсутствуют необратимые повреждения, и сократимость может восстанавливаться сразу после восстановления адекватного кровотока. В основе этого феномена лежит снижение сократительной активности миокарда в условиях хронической гипоперфузии с соответствующим балансом между низкой доставкой кислорода к миокарду и низкой его активностью (миокард как бы находится в "спячке").
Концепции "оглушенного" и "спящего" миокарда имеют свое клиническое приложение. Например, если у пациента с острым инфарктом миокарда или хронической ишемической болезнью сердца выявляются зоны потенциально обратимого нарушения локальной сократимости, это может повлиять на решение о целесообразности реваскуляризации миокарда, в частности путем операции аортокоронарного шунтирования. В настоящее время возможности выявления "оглушенного" и "спящего" миокарда ограничены, однако для этого применяются некоторые радионуклидные методики.
РЕМОДЕЛИРОВЛНИЕ ЛЕВОГО ЖЕЛУДОЧКА
После ИМ возникают изменения геометрии как поврежденной, так и интактной стенки левого желудочка. Изменения размеров камеры и толщины стенки влияют на долгосрочное состояние функции левого желудочка и прогноз.
В раннем постинфарктном периоде может произойти расширение зоны инфаркта, когда пораженный сегмент увеличивается в размерах без дополнительного некроза кардиомиоцитов. Расширение зоны инфаркта отражает истончение и дилатацию некротической ткани, видимо, за счет "соскальзывания" миофибрилл, в результате чего объем кардиомиоцитов в зоне инфаркта уменьшается. Расширение инфаркта может иметь негативные последствия, поскольку при этом увеличивается размер желудочка, а значит: 1) растет напряжение стенки, 2) нарушается систолическая сократительная функция, 3) повышается вероятность развития аневризмы.
Помимо раннего расширения зоны инфаркта, ремоделирование желудочка может включать дилатацию перегруженных неповрежденных сегментов, которые подвергаются повышенному напряжению стенки. Эта дилатация начинается в раннем постинфарктном периоде и затем продолжается в течение недель и месяцев. В начале дилатация полостей играет компенсаторную роль (она увеличивает сердечный выброс по закону Франка Старлинга), но прогрессирующее расширение желудочка может в конце концов привести к сердечной недостаточности и желудочковым аритмиям.
Неблагоприятное ремоделирование левого желудочка может быть предотвращено определенными вмешательствами. Например, в остром периоде инфаркта реперфузионная терапия позволяет ограничить размер инфаркта и, таким образом, уменьшить вероятность его расширения. Установлено также, что применение ингибиторов ангиотензин-превращающего фермента препятствует ремоделированию и снижает кратко- и долгосрочную смертность у постинфарктных больных.
Инфаркты с Q- и без Q-зубца
Патоморфологически, как мы уже видели, инфаркты миокарда делятся на трансмуральные и субэндокардиальные. Ранее считалось правилом, что при трансмуральных инфарктах на ЭКГ после исходной элевации сегмента ST образуется патологический зубец Q, в то время как для субэндокардиальных характерна депрессия сегмента ST без последующего появления зубца Q. Теперь, однако, известно, что изменения ЭКГ не столь четко коррелируют с патанатомической картиной, и между этими двумя типами инфарктов имеется значительное количество промежуточных форм. В частности, у некоторых пациентов с трансмуральным инфарктом на ЭКГ отсутствуют Q-зубцы, в то время как при субэндокардиальных инфарктах, напротив, зубцы Q иногда присутствуют. В целом, однако, инфаркты без Q-зубца обычно отличаются от инфарктов с Q-зубцом меньшими размерами и вызываются неокклюзирующими тромбами (или более короткими периодами тяжелой ишемии).