Курсовая работа (т): Генетика и биохимия алкоголизма
Таким образом, влечение к алкоголю и возможность
формирования алкогольной зависимости обусловлены гипофункцией
норадренергической, ГАМК-ергической, серотонинергической и опиоидной систем и
поддерживаются положительным подкреплением со стороны этих систем при приеме
этанола. Кратковременное введение этанола стимулирует функции этих
нейромедиаторных систем, а хроническое потребление истощает их и снижает
чувствительность адренорецепторов и опиоидных рецепторов. Эксперименты,
выполненные на генетических линиях животных, предпочитающих и отвергающих
этанол, позволяют предположить, что не только разная активность
этанолокисляющих ферментных систем, но и указанные выше различия в
нейрохимических структурах этих животных играют существенную роль в
наследственной предрасположенности к алкоголизму.
2.2 Метаболизм алкоголя в печени
Катаболизм этилового спирта осуществляется главным образом в печени. Здесь окисляется от 75% до 98% введённого в организм этанола.
Окисление алкоголя - сложный биохимический процесс, в который вовлекаются основные метаболические процессы клетки. Превращение этанола в печени осуществляется тремя путями с образованием токсического метаболита - ацетальдегида (рис. 6).
А. Окисление этанола NAD-зависимой
алкогольдегидрогеназой
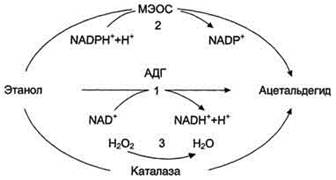
Рис. 6. Метаболизм этанола: 1 -
окисление этанола NAD+ - зависимой алкогольдегидрогеназой (АДГ); 2 - МЭОС -
микросомальная этанолокисляющая система; 3 - окисление этанола каталазой.
Алкогольдегидрогеназа катализирует обратимую реакцию, направление которой зависит от концентрации ацетальдегида и соотношения NADH/NAD+ в клетке.
Фермент алкогольдегидрогеназа -
димер, состоящий из идентичных или близких по первичной структуре полипептидных
цепей, кодируемых аллелями одного гена. Существуют 3 изоформы
алкогольдегидрогеназы (АДГ): АДГ1, АДГ2, АДГ3,
различающиеся по строению протомеров, локализации и активности (см. рис.7). Для
европейцев характерно присутствие изоформ АДГ1 и АДГ3. У некоторых
восточных народов преобладает изоформа АДГ2, характеризующаяся
высокой активностью, это может быть причиной их повышенной чувствительности к
алкоголю. При хроническом алкоголизме количество фермента в печени не
увеличивается, т.е. он не является индуцируемым ферментом.

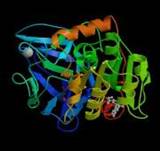
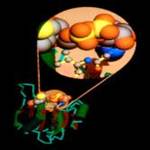
Рис. 7. Алкогольдегидрогеназа (АДГ)
Молекула представляет собой димер, (1) состоящий из двух одинаковых или различных цепочек (2). На рисунке (3) показан активный центр при взаимодействии с молекулами алкоголя
Б. Окисление этанола при участии цитохром Р450 - зависимой микросомальной этанолокисляющей системы системы
Цитохром Р450-зависимая
микросомальная этанолокисляющая система (МЭОС) локализована в мембране гладкого
эндоплазматического ретикулума гепатоцитов (ЭР) гепатоцитов. МЭОС играет
незначительную роль в метаболизме небольших количеств алкоголя, но индуцируется
этанолом, другими спиртами, лекарствами типа барбитуратов и приобретает
существенное значение при злоупотреблении этими веществами. Этот путь окисления
этанола происходит при участии одной из изоформ Р450 - изофермента Р450 II E1.
При хроническом алкоголизме окисление этанола ускоряется на 50-70% за счёт
гипертрофии ЭР и индукции цитохрома Р450 II E1.
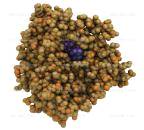
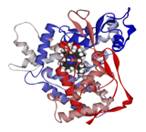
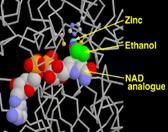
Рис. 8.
Система цитохрома P450
Участвует в окислении многочисленных соединений, как эндогенных, так и экзогенных. На рисунках показан внешний вид молекулы, схематическое изображение и процесс взаимодействия активного центра с молекулой алкоголя
В. Окисление этанола каталазой
Второстепенную роль в окислении этанола играет
каталаза, находящаяся в пероксисомах цитоплазмы (см. рис. 8) и митохондрий
клеток печени. Этот фермент расщепляет примерно 2% этанола, но при этом
утилизирует пероксид водорода.
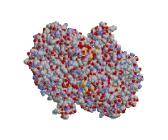
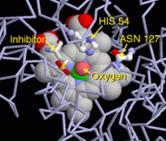
Рис 9. Каталазы - белки-антиоксиданты, которые
катализируют превращение перекиси водорода в воду и молекулярный кислород
На рисунке показан внешний вид, строение и механизм взаимодействия молекулы каталазы.
Г. Метаболизм и токсичность ацетальдегида
Ацетальдегид, образовавшийся из этанола, окисляется до уксусной кислоты двумя ферментами: FAD -зависимой альдегидоксидазой и NAD+ -зависимой ацетальдегиддегидрогеназой (АлДГ).
Повышение концентрации ацетальдегида в клетке вызывает индукцию фермента альдегидоксидазы. В ходе реакции образуются уксусная кислота, пероксид водорода и другие активные формы кислорода, что приводит к активации перекисного окисления липидов (ПОЛ).
Другой фермент ацетальдегиддегидрогеназа (АлДГ) окисляет субстрат при участии кофермента NAD+.
Полученная в ходе реакции уксусная кислота активируется под действием фермента ацетил-КоА-синтетазы. Реакция протекает с использованием кофермента А и молекулы АТФ. Образовавшийся ацетил-КоА, в зависимости от соотношения АТФ/АДФ и концентрации окса-лоацетата в митохондриях гепатоцитов, может "сгорать" в ЦТК, идти на синтез жирных кислот или кетоновых тел.
В разных тканях организма человека встречаются полиморфные варианты АлДГ. Они характеризуются широкой субстратной специфичностью, разным распределением по клеткам тканей (почки, эпителий, слизистая оболочка желудка и кишечника) и в компартментах клетки. Например, изоформа АлДГ, локализованная в митохондриях гепатоцитов, обладает более высоким сродством к ацетальдегиду, чем цитозольная форма фермента.
Ферменты, участвующие в окислении этанола, - алкогольдегидрогеназа и АлДГ по разному распределены: в цитозоле - 80%/20% и митохондриях - 20%/80%. При поступлении больших доз алкоголя (более 2 г/кг) из-за разных скоростей окисления этанола и ацетальдегида в цитозоле резко повышается концентрация последнего. Ацетальдегид - очень реакционно-способное соединение; он неферментативно может ацетилировать SH-, NH2- группы белков и других соединений в клетке и нарушать их функции. В модифицированных (ацетилированных) белках могут возникать "сшивки", нехарактерные для нативной структуры (например, в белках межклеточного матрикса - эластине и коллагене, некоторых белках хроматина и липопротеинов, формирующихся в печени).
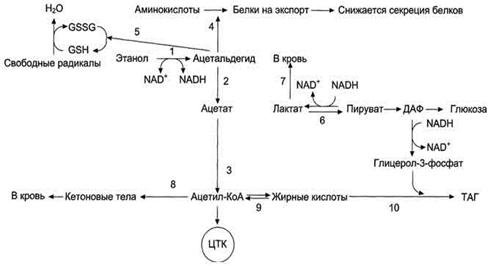
Рис. 10. Эффекты этанола в печени
1 2 3 - окисление этанола до ацетата
и превращение его в ацетил-КоА (1 - реакция катализируется
алкогольдегидрогеназой, 2 - реакция катализируется АлДГ). Скорость образования
ацетальдегида (1) часто при приёме большого количества алкоголя выше, чем
скорость его окисления (2), поэтому ацетальальдегид накапливается и оказывает
влияние на синтез белков (4), ингибируя его, а также понижает концентрацию
восстановленного глутатиона (5), в результате чего активируется ПОЛ. Скорость
глюконеогенеза (6) снижается, так как высокая концентрация NADH, образованного
в реакциях окисления этанола (1, 2), ингибирует глюконеогенез (6). Лактат выделяется
в кровь (7), и развивается лактоацидоз. Увеличение концентрации NADH замедляет
скорость ЦТК; ацетил-КоА накапливается, активируется синтез кетоновых тел
(кетоз) (8). Окисление жирных кислот также замедляется (9), увеличивается
синтез жира (10), что приводит к ожирению печени и гипертриацилглицеролемии.
Ацетилирование ядерных, цитоплазматических ферментов и структурных белков приводит к снижению синтеза экспортируемых печенью в кровь белков, например альбумина, который, удерживая Na+, поддерживает коллоидно-осмотическое давление, а также участвует в транспорте многих гидрофобных веществ в крови. Нарушение функций альбумина в сочетании с повреждающим действием ацетальдегида на мембраны сопровождается поступлением в клетки по градиенту концентрации ионов натрия и воды, происходит осмотическое набухание этих клеток и нарушение их функций.
Активное окисление этанола и ацетальдегида приводит к увеличению отношения NADH/NAD+, что снижает активность NAD+-зависимых ферментов в цитозоле и менее значительно в митохондриях.
Восстановление дигидроксиацетонфосфата, промежуточного метаболита гликолиза и глюконеогенеза, приводит к снижению скорости глюконеогенеза. Образование глицерол-3-фосфата повышает вероятность синтеза жира в печени. Увеличение концентрации NADH по сравнению с NAD+ (NADH>NAD+) замедляет реакцию окисления лактата, увеличивается соотношение лактат/пируват и ещё больше снижается скорость глюконеогенеза. В крови возрастает концентрация лактата, это приводит к гиперлактацидемии и лактоацидозу.окисляется ферментом дыхательной цепи NADH-дегидрогеназой. Возникновение трансмембранного электрического потенциала на внутренней митохондриальной мембране не приводит к синтезу АТФ в полном объёме. Этому препятствует нарушение структуры внутренней мембраны митохондрий, вызванное мембранотропным действием этилового спирта и повреждающим действием ацетальдегида на мембраны.
Можно сказать, что ацетальдегид опосредованно активирует ПОЛ, так как связывая SH-группы глутатиона, он снижает количество активного (восстановленного) глутатиона в клетке, который необходим для функционирования фермента глутатионпероксидазы, участвующего в катаболизме Н2О2. Накопление свободных радикалов приводит к активации ПОЛ мембран и нарушению структуры липидного бислоя.
На начальных стадиях алкоголизма окисление ацетил-КоА в ЦТК - основной источник энергии для клетки. Избыток ацетил-КоА в составе цитрата выходит из митохондрий, и в цитоплазме начинается синтез жирных кислот. Этот процесс, помимо АТФ, требует участия NADPH, который образуется при окислении глюкозы в пентозофосфатном цикле.
Однако в период острой алкогольной интоксикации, несмотря на наличие большого количества ацетил-КоА, недостаток оксалоацетата снижает скорость образования цитрата. В этих условиях избыток ацетил-КоА идёт на синтез кетоновых тел, которые выходят в кровь. Повышение в крови концентрации лактата, ацетоуксусной кислоты и гидроксибутирата служит причиной метаболического ацидоза при алкогольной интоксикации.
Как уже было сказано ранее, реакция
образования ацетальдегида из этанола протекает под действием
алкогольдегидрогеназы. Поэтому при повышении концентрации ацетальдегида и NADH
в клетках печени направление реакции меняется - образуется этанол. Этанол -
мембранотропное соединение, он растворяется в липидном бислое мембран и
нарушает их функции. Это негативно отражается на трансмембранном переносе
веществ, межклеточных контактах, взаимодействиях рецепторов клетки с
сигнальными молекулами. Этанол может проходить через мембраны в межклеточное
пространство и кровь и далее в любую клетку организма.
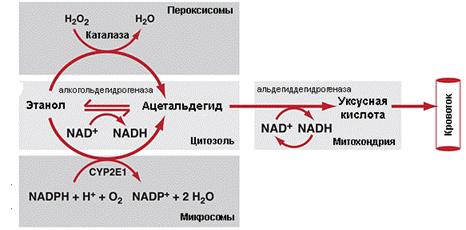
Рис. 11 Общая схема метаболизма
алкоголя в печени
Заключение
Для ученых наследственный характер алкоголизма означает, что в его основе лежит генетический элемент, передающийся от поколения к поколению. За последнее десятилетие достигнут значительный прогресс в идентификации генов и установлении их функций, что позволяет выявить генетическую подоплеку таких сложных заболеваний, как наркомания, токсикомания и алкоголизм. С помощью анализа малейших изменений в геноме индивидов из группы риска можно обнаружить специфические гены, которые влияют на физиологию человека и на риск развития у него определенного заболевания.
В настоящее время обнаружено около дюжины генов, которые вносят свой вклад в развитие алкоголизма. Многие из них способны провоцировать неадекватность поведения, депрессию, влиять на склонность к злоупотреблению спиртным, вызывать состояние тревоги и др. Таким образом, идентификация всех генов, обусловливающих реакцию организма на спиртное, поможет установить генетическую подоплеку не только алкоголизма, но и более широкого спектра патологий, а также даст возможность подобрать оптимальные способы лечения.
Американские ученые выяснили молекулярный механизм действия алкоголя на клетки мозга. Оказалось, что этиловый спирт связывается с калиевыми каналами нейронов, способствуя их открытию, что снижает активность клеток.
Через открытый GIRK ионы калия выходят из нейронов в межклеточное пространство, что подавляет активность этих клеток. Каналы GIRK обнаружены в различных нейронах, они могут опосредованно (рецепторно) активироваться ацетилхолином, норадреналином, допамином, опиатами и многими другими нейромедиаторами (“сигнальными молекулами”).
Наибольшее значение, по-видимому, имеет действие алкоголя на калиевые каналы, связанные с рецепторами типа B к тормозному медиатору ГАМК (гамма-аминомасляной кислоте). Предыдущие исследования объясняли эффект этанола именно взаимодействием c ГАМКB-рецепторами в областях мозга, ответственных за формирование памяти, принятие решений и импульсивное поведение, а также за судорожную активность.
Открытие американских ученых открывает путь к созданию принципиально новых препаратов для лечения алкогольной зависимости и эпилепсии.
Можно заключить, что к настоящему времени
получены данные, частично раскрывающие патогенез алкоголизма, однако их
совокупность образует довольно противоречивую картину. Несмотря на очевидный
прогресс, наметившийся в последние годы в понимании механизмов воздействия
алкоголя на ЦНС, единой однозначной теории, объясняющей все аспекты
формирования алкогольной зависимости, пока не существует.
Список литературы
1. Боринская С.А., Ким А.А., Рубанович А.В., Янковский Н.К. «Влияние аллелей гена ADH1B и уровня образования на характер потребления алкоголя у российских мужчин». Acta Naturae, 2013, т. 5, №3 (18), с. 103-110.
2. «Основы биохимии Ленинджера». - Нельсон Д, Кокс М. в 3-х томах. 2011 г.
. «Алкоголизм. Крах белкового обмена». Рослый И.М. 2013 г.
. «Наркология. Руководство для врачей». 2-е издание. Шабанов П.Д. 2012 г.
. «Алкогольная болезнь. Поражение внутренних органов». Под редакцией академика РАН В.С. Моисеева. 2014 г. Второе издание.
. Бочков Н.П. Клиническая генетика: Учебник. - 2 изд. - М.: ГЭОТАР - МЕД, 2002.
. Боринская С.А. Гены алкоголизма / Химия и жизнь. - 2008 - №7. - С.40
. Арзуманов Ю., Наговицина И. Генетические аспекты алкоголизма / Русский медицинский журнал. - 2001. - Т. 5. - №14. - С. 3-8.