Материал: MasterPass _ Pharmacology in 7 Days for Medical Students

MECHANISMS OF ACTION
The difference between sulfonylureas and meglitinides is that the latter have a rapid onset and short duration of action as compared to the former. Thus they are particularly effective in increasing insulin levels immediately after meals, thus correcting post-prandial hyperglycemia. In order to avoid post-prandial hyperglycemia, these agents are given from 1 to 30 minutes before meals.
An advantage of meglitinides is that these agents lack sulphur in their structure. Thus they can be used in type-2 diabetics with sulphur or sulfonylurea allergy. Also, combination therapy of meglitinides and metformin has been shown to be better than monotherapy with either agent in achieving normoglycemia.
Biguanides
Biguanides produce their pharmacological effects by multiple mechanisms including:
1Reduced hepatic and renal gluconeogenesis. It is pertinent to mention here that increased glucose output by the liver is the main cause of hyperglycemia in type-2 diabetics.
2Slowing of glucose absorption from GIT with increase glucose to lactate conversion by enterocytes.
3Direct stimulation of glycolysis in tissues with increased glucose removal from the blood.
4Reduction in plasma glucagon levels.
5Another beneficial effect of these drugs is lowering the levels of ‘bad cholesterol’ (i.e. LDL and VLDL cholesterol) and rising that of ‘good cholesterol’ (i.e. HDL cholesterol). These effects become apparent after 4–6 weeks of continuous use.
6Biguanides cause anorexia → ↓ food intake → ↓ glucose level. These drugs are thus the preferred antidiabetics in overweight/obese diabetics.
Because of the above-mentioned multiple beneficial effects, biguanides are the only oral hypoglycemics proven to decrease cardiovascular mortality.
Thiazolidinediones (TZDs)
Effect on insulin resistance: TZDs act by decreasing insulin resistance. Their target receptor is called peroxisome proliferator-activated receptor-gamma (PPAR-γ). These receptors are found in liver, adipose tissue and skeletal muscles. PPAR-γ receptors are complex and modulate expression of genes involved in:
1Lipid and glucose metabolism.
2Insulin signal transduction.
3Adipocyte and other tissue differentiation.
4Secretion of fatty acids.
The net effect of PPAR-γ receptors stimulation is increased insulin sensitivity (or decreased insulin resistance) in liver, adipose tissue and skeletal muscles.
Since the mechanism of action of TZDs involves gene regulation, they have slow onset and offset of activity over weeks or even months.
Effects on adipose tissue and lipid levels
1In adipose tissue the drug promotes glucose uptake and utilisation.
2Both pioglitazone and rosiglitazone increase the level of good cholesterol (i.e. HDL cholesterol). The level of bad cholesterol (i.e. LDL cholesterol) is lowered
59

PHARMACOLOGY IN 7 DAYS FOR MEDICAL STUDENTS
by pioglitazone but raised by rosiglitazone. Besides this, another bad effect of rosiglitazone is that this drug causes oedema, especially when combined with insulin. Rosiglitazone-insulin combination is thus not recommended.
3TZDs regulate adipocyte apoptosis and differentiation in such a way that there occurs an expansion in the subcutaneous fat.
TZDs also have significant effects on vascular endothelium, immune system, ovaries and tumour cells.
α-glucosidase inhibitors
Drugs included: acarbose; miglitol.
α-glucosidase inhibitors are competitive inhibitors of membrane-bound α-glucosidases (sucrase, maltase, glycoamylase, dextranase) in the intestinal brush border. α-glucosidases cause hydrolysis of oligosaccharides to glucose and other sugars. By inhibiting this enzyme, these drugs delay the digestion and thus absorption of ingested starch and disaccharides thus avoiding postprandial hyperglycemia. In fact post-prandial glycemic excursions are lowered by as much as 45–60 mg/dl. Thus the need of increased post-prandial release of insulin to control postprandial hyperglycemia is lowered by these drugs – an insulin-sparing effect.
Differences b/w acarbose and miglitol:
1Difference in potency: Miglitol differs structurally as well as in binding affinity from acarbose and is six times more potent in inhibiting sucrase.
2Difference in α-glucosidases targeted: Miglitol alone inhibits isomaltase and β-glucosidases, which split β-linked sugars such as lactose. Similarly, acarbose alone inhibits α-amylase.
Colchicine
Colchicine primarily produces its anti-inflammatory effects by binding to an intracellular microtubular protein called tubulin in granulocytes and other motile cells causing its depolymerisation. This in turn causes inhibition of leukocyte migration to the site of inflammation (→ ↓ phagocytosis at the site of inflammation).
Other anti-inflammatory effects include:
ADecreased release of inflammatory mediators responsible for pain (e.g. lactic acid and lipoproteins). Colchicine is in fact known to alleviate the pain of acute gout within 12 hours.
BInhibition of synthesis and release of proinflammatory mediators (e.g. prostaglandins and leukotriene B4).
CBlockage of cellular division by binding to mitotic spindles.
Allopurinol
After oral administration, allopurinol is approximately 80% absorbed and has a terminal serum t½ of 1–2hrs. Allopurinol inhibits uric acid synthesis:
Hypoxanthine → xanthine → uric acid
Xanthine oxidase is the catalysing enzyme for both these steps.
Allopurinol is a pro-drug. It is metabolised by xanthine oxidase enzyme to alloxanthine. Alloxanthine in turn inhibits xanthine oxidase enzyme thus inhibiting uric acid synthesis and increasing the concentrations of hypoxanthine and xanthine. Alloxanthine has a long duration of action so that allopurinol needs to be given only once daily.
60
|
|
|
|
|
|
|
|
|
|
|
|
|
|
MECHANISMS OF ACTION |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purines |
|
Hypoxanthine |
|
|
|
|
Xanthine |
|
|
|
|
|
Uric acid |
|
|
|
Allantoin |
|||
(adenine & guanine) |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
Xanthine |
Xanthine |
|
|
Uricase |
||||||||||||||||
|
|
|||||||||||||||||||
|
|
|
||||||||||||||||||
|
|
oxidase |
oxidase |
|
|
|
|
Probenecid |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Allopurinol |
|
|
|
|
|
|
|
|
sulfinpyrazone |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
Excreted in urine
Figure 3.5 Purine metabolism and pharmacological interventions with allopurinol, probenecid and sulÞnpyrazone
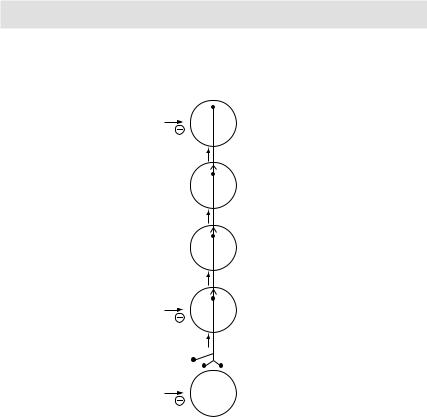
Morphine
Opioid receptors are present both in the ascending pathways responsible for pain transmission from the periphery to the higher centres and the descending pathways (in midbrain and medulla) responsible for pain modulation.
Action at post-synaptic level: agonistic action on Mu (μ) receptors with resultant opening up of K+ channels: Morphine binds to specific opioid receptors situated in the GIT, urinary
Presynaptic (primary sensory) neuron
Postsynaptic (secondary relay) neuron
|
µ-receptor |
|
|
||
↓Ca++ influx means |
|
|
|
|
Direction |
|
|
|
|
of |
|
↓neurotransmitters |
|
|
|
Ca++ |
|
|
|
|
|||
|
|
|
propagation |
||
release from the |
Ca++ |
|
of |
||
presynaptic neuron |
|
|
|
|
action |
into the synaptic cleft |
|
|
|
|
|
|
|
|
|
potential |
|
|
|
|
|
|
|
K+ µ-receptor
↓K+ efflux means 
 hyperpolarization
hyperpolarization
of the postsynaptic neuron
Figure 3.6 Mechanism of action of morphine (µ-receptor agonists)
61

PHARMACOLOGY IN 7 DAYS FOR MEDICAL STUDENTS
bladder and other smooth muscles as well as in CNS (brain and spinal cord including peri-aqueductal grey matter, nucleus rafimagnus, nucleus reticularis, paragigantocellularis, substantia gelatinosa, limbic system and in somatosensory cortical area). Mu
(μ) receptors have got great affinity for morphine. When morphine binds to these receptors, it causes opening up of K+ channels with resultant efflux of K+ ions and hyperpolarisation at postsynaptic neurons causing neuronal depression.
Action at pre-synaptic level: agonistic action on mu (μ), kappa (κ) and delta (δ) receptors with resultant closure of pre-synaptic, voltage-gated, Ca++ channels: Morphine acts on μ, κ, δ receptors found on the pre-synaptic terminals of the nociceptive primary afferents resulting in closure of the pre-synaptic, voltage-gated Ca++ channels present. Inhibition of Ca++ influx inhibits the release of neurotransmitters like acetylcholine, norepinephrine, serotonin, somatostatin, bombesin and substance P. These neurotransmitters mediate pain perception in the spinal cord. When their release into the synaptic cleft is inhibited, post-synaptic neuronal firing and thus transmission of nociception decreases.
Action at the periphery: Agonistic action on delta (δ) receptors with resultant depression of peripheral nociceptors: Morphine and other opioid analgesics also depress the discharge from the peripheral nociceptive afferent neurons. This in turn leads to impaired nociceptive input and thus impaired pain perception.
At molecular level, it is pertinent to mention here that all three opioid receptors (μ, κ, δ) are coupled to their effectors by G proteins, and activate phospholipase C or inhibit adenylyl cyclase.
Box 3.1 CNS effects of morphine
1Analgesia. Both the sensory and emotional aspects of pain experience are attenuated, i.e. although patients are still aware of the presence of pain, the sensation is not unpleasant.
2Sedation via κ-receptor activation. At higher doses, it can even cause stupor and coma.
3Euphoria. By stimulating ventral tegmentum, morphine produces a powerful sense of well-being and euphoria.
4Miosis (except mepridine, which has a muscarinic blocking action). Nalaxone (an opioid antagonist) and atropine can reverse miosis.
5Inhibition of respiratory centre in medulla oblongata via µ-receptor activation
with resultant decreased response to PCO2 (→ ↑ PCO2 → cerebrovascular dilatation → ↑ ICP).
6Nausea and vomiting (by activating chemoreceptor trigger zone). Anti-emetics are routinely given with morphine and other narcotic analgesic injections.
7Suppression of cough reßex (by an unknown mechanism).
Benzodiazepines (BDZs)
GABA (γ-aminobutyric acid) is the major inhibitory neurotransmitter in the CNS. Benzodiazepines are GABA-ergic (meaning they potentiate the post-synaptic inhibitory actions of GABA in various areas of the brain). GABA receptors are composed of α, β, and γ-subunits. Five or more combinations of these subunits constitute the different
62
MECHANISMS OF ACTION
Pain transmission from nociceptors to cerebral cortex: Since different painkillers act at different/multiple sites, a combination of them is more effective than monotherapy with any one analgesic agent
Opioids |
Cerebral |
|
cortex |
|
Thalamus |
|
Brain |
|
stem |
Opioids |
|
NSAIDs |
Spinal |
Antidepressants |
cord |
Anticonvulsants |
|
|
Free nerve endings |
NSAIDs |
Nociceptors |
Figure 3.7 Pain transmission from nociceptors to cerebral cortex
GABA receptor subtypes present on the post-synaptic membranes in the CNS. Depending upon the subtype activated, varying pharmacological effects of BDZs are produced (Table 3.1).
BDZs bind with ‘BDZ-binding sites’ located at the interface of α, and γ2-subunits. This leads to displacement of a substance GABA-modulin, which occupies GABA receptors and keeps them in a relatively non-functional state. This in turn increases the affinity GABA for its receptor (GABAA) leading to increased opening up of chloride channels, hyper-polarisation and thus depression of the post-synaptic neurons. In other words, BDZs increase the affinity GABA for its receptor, in turn increasing the frequency of chloride channels opening produced by GABA.
It is pertinent to mention here that BDZs are not GABA-mimetic (meaning they do not have effects like GABA) and the presence of GABA in the CNS is necessary for the actions of BDZs. If we inhibit the synthesis of GABA with thiosemicarbazide, BDZs become ineffective. Similarly if we block post-synaptic receptors of GABA with bicuculline, BDZs again will become ineffective.
63