Материал: Процессы гидрирования и дегидрирования органических соединений
Процессы гидрирования и дегидрирования органических соединений
Содержание
Введение
1. Понятие о катализаторах
2. Процесс гидрирования непредельных соединений
2.1 Традиционные процессы и катализаторы гидрирования
2.2 Современные тенденции в разработке и использовании новых
катализаторов гидрирования
2.3 Катализаторы на основе благородных металлов
2.4 Процесс гидрирования
3. Процесс дегидрирования органических веществ
3.1 Разновидности дегидрирующего действия катализаторов
3.2 Современные представления о природе активной поверхности
катализаторов дегидрирования
3.3 Процесс дегидрирования
Заключение
Список литературы
Введение
катализатор дегидрирование органический
Каталитическое гидрирование включает большую группу реакций присоединения водорода по ненасыщенным связям С=С, С=О, C≡N, С≡С и т.д., а также реакции гидрогенолиза (разрыва и восстановления) связей С-С, С-О, С-S и др.
Кроме гетерогенных катализаторов, применяющихся в крупнотоннажных процессах, используют также гомогенные металлокомплексные и гидридные катализаторы (преимущественно в тонком органическом синтезе). Расширяется применение энантиоселективных катализаторов гидрирования, в основном металлокомплексных соединений никеля, кобальта и других металлов с оптически активными лигандами.
Наиболее широкое распространение получили нанесенные никелевые катализаторы гидрирования, за ними идут катализаторы на основе благородных металлов. В процессах гидрооблагораживания и гидродесульфуризации широко применяются бинарные Ni-Мо-, Ni-W-, Со-Мо- и Со-W- катализаторы. Новыми перспективными катализаторами гидрирования являются карбиды металлов [1].
К реакциям каталитического дегидрирования способны углеводороды различных гомологических рядов (парафины, нафтены, олефины и циклоолефины, алкилароматические углеводороды). В результате реакции отщепляется водород и образуются более ненасыщенные соединения или происходит циклизация (дегидроциклизация). Катализаторами дегидрирования являются различные металлы и оксиды. Основные побочные реакции, протекающие при каталитическом дегидрировании: крекинг, изомеризация, коксообразование. Каталитическое дегидрирование является одним из важнейших промышленных процессов переработки углеводородного сырья [2].
Цель работы: рассмотреть процессы гидрирования и дегидрирования органических соединений, а так же природу катализаторов и механизмы их действия.
1. Понятие о катализаторах
Катализатором называется вещество, которое своим присутствием в небольшом количестве в смеси веществ способствует химической реакции между ними (определение Берцелиуса, 1836) или увеличивает скорость реакции (определение Оствальда, 1894). Второе определение основано на предположении, что реакция протекает и в отсутствие катализатора, но с очень малой, возможно неизмеримой, скоростью. Действительно, известны многочисленные реакции, протекающие в отсутствие катализатора, но в присутствии катализатора их скорость резко возрастает. Известно также большое число реакций, которые идут только в присутствии определенного катализатора. В некоторых случаях одни и те же реагирующие вещества при замене катализатора образуют другие продукты реакции. Надо полагать, что в последних случаях катализатор играет существенную роль, поскольку влияет на ход реакции, так что старое определение Берцелиуса, верно, по крайней мере, в отдельных случаях.
Следует подчеркнуть, что катализатором всегда является вещество. Было бы неправильно говорить, например, о каталитическом действии света.
Когда говорят, что катализатор не принимает участия в реакции, подразумевают, что после реакции он находится качественно и количественно в том же состоянии, что и до реакции. Поэтому катализатор не включают в уравнение химической реакции. Кроме того, нет необходимости использовать его в каком-либо определенном стехиометрическом соотношении с реагентами. Иногда достаточно ничтожно малого количества катализатора, чтобы вызвать реакцию.
Катализаторы ускоряют или вызывают только термодинамически возможные реакции, т.е. реакции, протекающие самопроизвольно с уменьшением свободной энергии в сторону установления равновесия.
Изучение реакций, протекающих как в присутствии, так и в отсутствие катализаторов, раскрыло важную сторону механизма их действия, которое заключается в том, что катализаторы уменьшают энергию активации реакций. Например, известно, что энергия активации гомогенной реакции в газовой фазе - разложения йодистого водорода 2HI → H2 + I2 - равна 44,3 ккал/моль. Энергия активации этой реакции, проводится в присутствии металлического золота в качестве катализатора (гетерогенный катализ), равна 25 ккал/моль, а в присутствии платины - только 14 ккал/моль.
Различают две большие группы каталитических реакций: гомогенный и гетерогенный катализ. В первой группе реакций катализатор и субстрат образуют одну фазу, обычно жидкую. Во второй группе реакций катализатор является твердым, а субстрат - жидким или газообразным. Третья группа каталитических реакций - ферментативные реакции - охватывает процессы, исключительно важные для животных и растительных организмов. Участвующие в этих реакциях катализаторы - ферменты - являются органическими соединениями (белки) с огромными молекулами (макромолекулы), вырабатываемыми живыми клетками.
Гомогенный катализ. Катализаторами огромного большинства реакций этой группы являются кислоты и основания. В настоящее время в результате обстоятельных кинетических исследований установлено, что в реакциях, катализируемых кислотами, субстрат ведёт себя как основание, а промежуточный продукт является его сопряженной кислотой. В реакциях, катализируемых основаниями, субстрат ведет себя как кислота, отдавая катализатору протон. После каждой элементарной реакции катализатор - кислота или основание - регенерируется. Большинство этих реакций относится к области органической химии.
Ферментативными реакциями являются реакции гидролиза, гидрирования и дегидрирования (присоединение водорода к молекулам субстрата или отщепление водорода от его молекул), реакции окисления (перенос электронов), реакции переноса групп фосфорной кислоты и органических групп. Резко выраженная специфичность ферментов указывает на то, что между ферментом и молекулой субстрата образуются химические соединения, благодаря которым последние активируются (теория Михаэлиса и Ментен, 1913).
Уравнение
Михаэлиса-Ментен: основное уравнение ферментативной
<#"822644.files/image001.gif">
Уравнение
имеет вид:
![]()
где
![]()
![]() -
максимальная скорость реакции, равная kcatE0;
-
максимальная скорость реакции, равная kcatE0; ![]()
![]() -
константа Михаэлиса, равная концентрации субстрата, при которой скорость
реакции составляет половину от максимальной; S - концентрация
субстрата [3].
-
константа Михаэлиса, равная концентрации субстрата, при которой скорость
реакции составляет половину от максимальной; S - концентрация
субстрата [3].
Гетерогенный катализ имеет широкое и важное применение в промышленности. Гетерогенный катализ особенно часто используется в реакциях окисления (пример: 2SO2 + O2 → 2SO3), в реакциях восстановления или гидрирования (пример: N2 + ЗН2 → 2NH3), в реакциях дегидрирования (осуществляемых в нефтехимической промышленности с использованием в качестве катализаторов платины, оксиды хрома или сульфида молибдена) и в реакциях разрыва молекул углеводородов нефти (крекинг) в присутствии катализаторов кислотного характера (оксиды алюминия, алюмосиликаты).
Твердые катализаторы тем активнее, чем больше их поверхность, т.е. чем они больше измельчены, и чем ниже температура их получения и последующей обработки. Так, катализаторы из металлического никеля, используемые в реакциях гидрирования, представляют собой пирофорные порошки, полученные восстановлением оксида никеля NiO водородом примерно при 300 °С. При нагревании примерно до 700 °С эти катализаторы полностью теряют активность, поскольку атомы на их поверхности перестраиваются в более устойчивое состояние. Можно сделать вывод, что в реакции принимает участие не вся поверхность катализатора, а только определенные активные центры (Тейлор, 1926).
Давно известно, что химическая реакция при гетерогенном катализе происходит во внешнем слое молекул, адсорбированных на поверхности катализатора (Лэнгмюр, 1916) или точнее на активных центрах катализатора. Различают два типа адсорбции: адсорбцию вандерваальсовыми силами при низкой температуре и активированную адсорбцию, или хемосорбцию. Последний процесс, единственно важный при гетерогенном катализе, состоит в образовании химических соединений между молекулами реагентов и поверхностными атомами катализатора. Теплоты, выделяющиеся при вандерваальсовой адсорбции, малы (около 4 ккал/моль для таких газов, как O2, СО и N2 на Сr2O3 при -183 °С), но достигают значения обычных теплот реакции при хемосорбции (50 ккал/моль для O2 на Сr2O3 при 0°С). Вандерваальсова адсорбция осуществляется мгновенно, тогда как хемосорбция происходит медленно, но ее скорость возрастает с температурой.
В обычной каталитической реакции продукты реакции десорбируются с катализатора и диффундируют в массу газа (или жидкости), а их место тотчас же занимают молекулы реагентов, которые повторяют тот же элементарный процесс. Таким образом, один активный центр поверхности катализатора превращает большое количество молекул реагента. Некоторые вещества, например сероводород H2S, фосфин Н3Р и различные соединения азота, прочно закрепляются на поверхности катализаторов (металлов), блокируя активные центры и тем самым замедляя каталитический процесс (отравление катализаторов) [4].
2. Процесс гидрирования непредельных соединений
Гидрирование или гидрогенизация (от позднелатинского hydrogenium - водород), деструктивная гидрогенизация, - совокупность химических процессов, происходящих при воздействии водорода на органическое вещество. В топливоперерабатывающей промышленности гидрогенизацию применяют для получения из твёрдых горючих ископаемых (угли, сланцы), а также низкосернистых нефтей и тяжёлых нефтяных остатков моторного горючего, смазочных масел и химических продуктов. Гидрогенизация твёрдого топлива является универсальным методом получения из него синтетического жидкого топлива. Также важный резерв для замены сырой нефти горючими сланцами, битумами, углями [5,6].
Развитие исследований в области гидрогенизации относится к 1897-1900 гг., когда П. Сабатье (Франция) и Н.Д. Зелинский (Россия) со своими учениками разработали основы гидрогенизации катализа органических соединений. Влияние давления водорода на ускорение реакций гидрогенизации органических соединений было установлено вначале XX в. В.Н. Ипатьевым. Промышленное применение гидрогенизации твёрдого топлива впервые было получено в 30-40-х гг., в Германии. Перед 2-й мировой войной (1939-1945) установки по гидрогенизации угля и угольных смол работали также в Великобритании, Италии, Корее; в СССР были построены два опытных завода. В послевоенный период в основе переработки нефти сырья применяли гидрогенизацию. Начиная с 60-х гг. ведутся работы по гидрогенизации твёрдого топлива с целью создания экономически эффективных процессов производства синтетических жидких топлив.
В СССР был разработан процесс гидрогенизации угля для получения моторного
горючего, котельного топлива и химикатов. Процесс осуществляется при температуре
420-430 °С, давлении водорода 10 Па, в присутствии активных катализаторов,
растворителя и органических добавок-ингибиторов реакций радикальной
полимеризации. В зависимости от исходного сырья выход жидких продуктов 85-95%
(технологическая схема процесса дана на рис.1.).
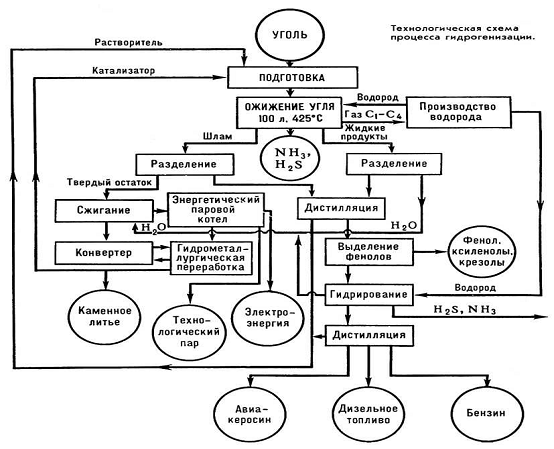
Рис.1. Процесс гидрогенизации угля для получения моторного горючего,
котельного топлива и химикатов.
В США, Великобритании, ФРГ разрабатывается ряд процессов по
гидрогенизации угля с катализатором и без катализатора, под давлением водорода
1-7 и 15-30 Па, температура 400-500 °С, а также экстракции угля растворителями
с последующей гидрогенизации экстрактов. Исследования по гидрогенизации
твёрдого топлива и тяжёлых нефти остатков ведутся в Японии, Индии, Австралии,
Польше и др. [3,5].
2.1 Традиционные процессы и катализаторы гидрирования
В первом промышленном процессе гидрирования использовался никелевый катализатор, открытый Сабатье в 1897 г. Позже были разработаны другие известные катализаторы гидрирования: металлы VI-VIII групп, в том числе металлы платиновой группы, на носителях и в составе сложных оксидных систем, а также скелетные металлические катализаторы.
Результаты исследования Ипатьевым в начале XX в. каталитических реакций гидрирования под давлением послужили основой для создания процессов, широко используемых в настоящее время в промышленности. Был разработан ряд специфических катализаторов гидроочистки, получаемых пропиткой или соосаждением (Аl-Со-Мо-, Ni-Мо-, сульфидные Ni-W-S- катализаторы и т.д.), которые устойчивы к каталитическим ядам, содержащимся в нефтяном сырье (S-, N-, О- содержащие соединения).
Объем производства катализаторов для получения органических соединений не очень велик. К числу крупнотоннажных относятся промышленные процессы гидрирования жиров, 2-этилгексеналя, ацетилена (для удаления его из этилена), бензола в циклогексан в производстве капролактама и некоторые другие. Расширяется применение катализаторов гидрирования при производстве лекарств и физиологически активных соединений. Важными показателями для катализаторов этой категории являются высокая селективность, длительный срок службы, соответствие экологическим требованиям. В основном такими характеристиками обладают металлические (Ni, Pt, Pd, Rh, Ru) катализаторы на носителях, медно-хромовые и никель-хромовые соосажденные катализаторы.
На примере гидрирования бензола в циклогексан можно проследить тенденцию к использованию того или иного катализатора в зависимости от того, насколько жесткие требования предъявляются к чистоте конечного продукта.
С начала века гидрирование бензола традиционно осуществляли на никеле (никель Ренея, пропиточные катализаторы) и платиновых металлах на носителях. В ранних промышленных процессах, проводившихся при повышенных температуре и давлении, наряду с основной протекали и побочные реакции: крекинг с образованием n-гексана, изомеризация циклогексана в метилциклопентан, изомеризация и крекинг с выделением более легких углеводородов.
Наилучшие результаты при получении высокочистого циклогексана обеспечивает двухстадийная схема: на первой стадии используют нанесенный Ni-катализатор (30-45% Ni/Аl2О3), а на второй - Pt-катализатор на носителе (0,2-1,0% Pt/Аl2О3). Чистота циклогексана достигает при этом 99,57-99,88%.
Большое внимание в нашей стране уделяли соосажденным никель-хромовым катализаторам, обеспечивающим получение высокочистого циклогексана в трубчатых реакторах в несколько стадий при строгом соблюдении технологии. Катализаторы готовили осаждением азотнокислых солей Ni и Cr раствором соды при комнатной температуре. Затем следовали фильтрация, промывка осадка от нитрат-ионов, сушка, прокаливание, таблетирование и активация в токе водорода. Содержание Ni составляло 48-63%, а Сr2О3 - 37-52%. Недостатками этого и других способов получения Ni-Cr-катализаторов являются многостадийность, громоздкость фильтровального оборудования, большие объемы сточных вод на стадии промывки и т.д.
В связи с этим был предложен бессточный метод получения Ni-Сr-катализаторов, включающий стадию смешения растворов Ni(NO3)2 и (NH4)2Gr2O7 с последующими сушкой и прокаливанием смеси солей путем ее распыления на нагретую насадку из инертного материала в режиме кипящего слоя при 310-340 °С, таблетированием и восстановлением [1].