Материал: Приготовление нанесенных катализаторов и их химический анализ
Значение рН золя также оказывает существенное влияние на величину поверхности силикагеля. При повышении рН золя кривая удельной поверхности также проходит через максимум, положение которого зависит от концентрации SiO2: для серии А (концентрация золя 1.8 моль SiO2/л) максимум лежит при рН около 2, а для серии Б (концентрация SiO2 0,83 моль/л) - при рН около 3.5 при увеличении рН выше значений соответствующих максимальной поверхности наблюдается довольно резкое уменьшение величины поверхности от 700-800 до 200 м2/г.
Влияние продолжительности созревания. Большое влияние на величину поверхности формирующегося ксерогеля оказывает степень старения (глубина синерезиса). Уже давно показано [11], что гель, полученный из сильнокислого золя и не претерпевший синерезиса, обладает меньшей поверхностью, чем гель после синерезирса. Влияние продолжительности старения на величину поверхности силикагеля зависит от условий приготовления геля. Например, при увеличении продолжительности старения до 120 часов поверхность образцов, приготовленных при рН 6,5 остается постоянной, равной 700 м2/г.
Влияние состава промывной жидкости и пропитки гидрогелей на величину поверхности силикагелей было предметом многочисленных исследований Ней Марка с сотр. [12,13], которые обнаружили большое влияние рН промывной воды. Силикагели, промытые подкисленной водой, не зависимо от рН золя обладает поверхностью около 600 м2/г. Гели, промытые при рН 10, имеют поверхность около 400 м2/г. Поверхностью приблизительно 300 м2/г обладают гели, промытые водопроводной водой. Увеличение поверхности силикагеля, полученного в щелочной среде, при промывке подкисленной водой объяснено обменом катионов натрия на ионы водорода; уменьшение поверхности силикагеля полученного в кислой среде, при промывке водопроводной водой объяснено обменом ионов водорода на катионы кальция.
Влияние природы адсорбированных катионов на величину поверхности ксерогеля SiO2 исследовано в работе Высоцкого и Шаля [14]. Гель, полученный при рН 4 промывали раствором соляной кислоты и растворами бикарбонатов или хлоридов натрия, кальция и калия, а затем дистиллированной водой. Найдено, что удельная поверхность силикагеля определяется природой катионов, а именно: уменьшается в ряду Н+, Са2+, Na+, K+. Уменьшение поверхности силикагеля под влиянием катионов авторы связывают с понижением гидрофильности частиц гидрогеля в местах адсорбции, что способствует их агрегации. Влияние природы катиона на величину удельной поверхности связывается с теплотой гидратации.
Температура промывки и старения также оказывает большое влияние на формирование поверхности силикагеля: с увеличением температуры величина поверхности уменьшается. Так, в работе Вольфа и Байера [15], было показано, что при увеличении температуры промывных вод от 50 до 100 ˚С величина поверхности уменьшается от 618 до 320 м2/г. Особенно резко это влияние проявляется при температурах выше 100 ˚С в гидротермальных условиях.
Механизм формирования поверхностей силикагелей
Изложенный выше материал позволяет сделать вывод, что для силикагелей, приготовленных описанными методами, наблюдаются сходные закономерности формирования поверхности:
а) при получении гидрогелей в условиях низких значений рН и температур образуются ксерогели с высокоразвитой поверхностью;
б) при повышении рН, продолжительности промывки и старения, а в ряде случаев и температуры величина поверхности уменьшается, особенно резко особенно в условиях гидротермальной обработки;
в) включение в состав геля катионов вследствие неполного гидролиза или в результате хемосорбции из маточного раствора обусловливает уменьшение размера поверхности.
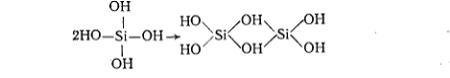
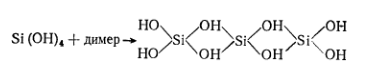
Работами ряда авторов [16], показано, что при введении
щелочного силиката в кислоту возникает мономерная кремневая кислота, которая с
течением времени конденсируется, образуя молекулы поликремневых кислот:
Относительно порядка присоединения молекул мономера, мнения различных авторов расходятся: некоторые предполагают, что конденсация идет по концевым группам вследствие чего, на начальной стадии процесса полимолекула имеет линейное строение, другие считают, что конденсация протекает во всех трех направлениях. Однако, независимо от начального механизма частицы золя, образующегося в результате конденсации, имеют гигроскопические исследования, позволяющие непосредственно наблюдать образование и поведение частиц в золе [17]. На основании этих исследований можно сделать вывод, что внутренняя часть глобулы состоит из компактной SiO2, а внешняя оболочка представлена двойным слоем, состоящим из ионов металла, гидроксильных групп и молекул воды.
Золь кремнекислоты неустойчив: с течением времени происходит дальнейшая агрегация глобул, вязкость возрастает и по истечении некоторого времени золь превращается в гель.
Согласно работе [18], имеет место следующий механизм
конденсации кремневой кислоты: при рН меньше 2, когда существует динамическое
равновесие между Н+ в растворе и группами ОН, на поверхности
макромолекул SiO2 образуется положительно заряженные группы:
![]()
которые могут реагировать с мономерными молекулами кремневой
кислоты:
![]()
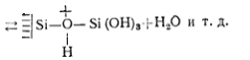
При рН больше 2 поверхность частиц несет отрицательный заряд
и может реагировать со следующими мономерными молекулами:
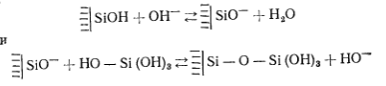
Автор считает, что так как величина поверхности силикагеля определяется размером частиц, из которых он построен, то она должна зависит от скорости и продолжительности конденсации. Поскольку при рН около 2 скорость конденсации мала, удельная поверхность силикагеля велика. При более высоких рН, скорость конденсации возрастает и соответственно снижается величина поверхности.
Если бы эти представления были справедливы, то повышения рН при осаждении, созревании и промывки приводило бы к необратимым изменениям величины поверхности. Однако, исследование [10,11,12] показали, что при снижении рН в зависимости от возраста геля происходит более или менее полное восстановление первоначальной величины поверхности. Только в случае геля, старевшего в течении длительного времени наблюдается существенная необратимость изменения поверхности.
Окончательное формирование поверхности силикагеля в основном происходит в процессе сушки, когда под действием мощных поверхностных сил, развивающихся при обезвоживании, происходит сближение глобул между собой. В результате становится возможным взаимодействия поверхностных гидроксильных групп с образованием химических связей, что и обусловливает необратимость изменений величины поверхности высушенных образцов. При этом возможны два случая: размер первичных частиц может остаться неизменным, как это наблюдалось в работе Киселева и соавт. [19], либо может существенно увеличиваться в результате коалесценции двух или нескольких частиц.
Механизм формирования поверхности гидроокисей
Дисперсность и величина поверхностей гидроокисей могут сильно варьировать в зависимости от их природы и условий приготовления. Даже для одного и того же вещества при изменении рН, температуры осаждения и других факторов величина поверхности может изменяться более чем на 2 порядка.
Рассмотрим более подробно протекание отдельных стадий процесса формирования гидроокисей.
Гидролиз. Первая стадия образования гидроокиси - гидролиз исходного вещества. Установлено, что уже при растворении соли в воде происходит быстрая гидратация катиона. С течением времени или повышением рН происходит гидролиз гидратированной частицы, при котором имеет место перенос протона по связи Ме-ОН2. Результатом этого является замещение аква группы внутри комплексного иона на гидроксил и освобождение протона. Продукты гидролиза, образовавшиеся на этой стадии процесса, конденсируются. Состав продуктов зависит как от условий образования (способа получения, рН среды), так и от природы катиона.
Конденсация. В результате полимеризации и конденсации первичных продуктов гидролиза образуются полимолекулы, которые наряду с гидроокисью могут содержать большее или меньшее количество основных (или кислых) солей. Возникающие в результате этого сравнительно крупные макромолекулы являются первичными частицами золя. Во многих случаях они имеют сферическую форму. Можно было предполагать, что для большинства гидроокисей это действительно имеет место, что частицы золя и являются первичными элементами будущей структуры ксерогеля.
Известно что, процесс образования новой фазы состоит из двух основных стадий: образование зародыша и его роста. Размер частиц определяется соотношением скоростей этих стадий.
Коагуляция. Образовавшиеся на предыдущей стадии высокодисперсные системы - золи в принципе неустойчивы и с течением времени в зависимости от условий образований и природы гидроокиси более или менее быстро коагулируют. Процесс коагуляции протекает ступенчато: в результате столкновений из первичных частиц возникают агрегаты, имеющие форму "цепей" и "гроздей" [20], в которых частицы объединены слабыми коагуляционными силами. Размеры рыхлых агрегатов первичных частиц, достигающая порядка сотен ангстрем, зависит от химической природы вещества, а для одного и того же вещества от условий образования (концентрации, рН и т.д.). В зависимости от условий получений-температуры, рН и наличия коагуляторов в среде - вторичные образования ("цепи" и "гроздья") постепенно объединяются в крупные хлопья, которые со временем теряют агрегативную устойчивость и коагулируют. В отличие от первичных частиц величина агрегатов в зависимости от условий получений и возраста систем может колебаться в широких пределах: в ряде работ показано, что различные гидроокиси образую агрегаты от 200 до 1000 ангстрем [21]. Коагуляция приводит к образованию геля (студня) или коагеля. Коагуляционные связи непрочны, и образовавшийся гель или осадок при изменении условий, например механическом воздействии или введении пептизатора, может снова превратиться в золь. Однако по мере старения геля реакционно способные группы, находящиеся на поверхности различных частиц, могут реагировать между собой с выделением воды и образованием кислородных мостиков между частицы. Возникающие, в результате этого "конденсационные структуры" значительно прочнее коагуляционных, и происходящие при их образовании изменения свойств геля необратимы.
Стадия коагуляции оказывает существенное влияние на процесс приготовления катализатора или носителя, так как от нее зависят физические свойства осадка - его объем, количество связываемой воды, фильтруемость и прочее.: чем лучше скоагулирован осадок, т.е. чем крупнее и плотнее образовавшиеся агрегаты, тем легче протекают фильтрации и отмывка продукта.
Переконденсация и коалесценция. Наряду с образованием крупных агрегатов, происходящим без изменения размеров первичных частиц, понижение свободной энергии системы может осуществляться в результате роста первичных образований. Это возможно благодаря процессам переконденсации и коалесценции. Причина переконденсации заключается в более высокой растворимости высокодисперсных частиц по сравнению с грубодисперсными, вследствие чего при подходящих условиях происходит растворение мелких частиц с последующим выделением вещества на более крупных. По аналогии с известным изменением дисперсности жидких капель этот процесс обычно называют "перегонкой" или "переконденсацией". Протеканию процесса по этому механизму благоприятствует высокая концентрация исходного вещества в растворе, и при повышении температуры и растворимости доля этого механизма возрастает.
Более распространенным процессом, приводящим к уменьшению поверхности, является коалесценция первичных частиц во время сушки гидрогеля. Глубина протекания процесса коалесценции в основном зависит от степени гидролиза исходных веществ: при малом содержании продуктов неполного гидролиза коалесценция незначительна и размер частиц близок к минимальному. С увеличением содержания основных (или кислых) солей размер частиц возрастает, а величина поверхности уменьшается.
Кристаллизация и старение. Если гидроокись способна
кристаллизоваться, то скорость и глубина кристаллизации могут оказать
существенное влияние на формирование поверхности ксерогеля. Наиболее четко
проявляется это влияние в случае медленно кристаллизующихся и склонных к
фазовым превращениям гидроокисей, для которых возможны измерения скоростей всех
стадий. Типичные представители этого типа - гидроокиси алюминия, железа, хрома,
меди и др. Многими авторами показано, что в большинстве случаев свежеосажденные
гидроокиси этого типа не имеют определенного химического состава, содержат
большие или меньшие количества сверхстехнометрической "воды" и
продуктов неполного гидролиза. Различными методами установлено, что не
старевшие гидроокиси состоят из сравнительно крупных бесформенных агрегатов
более мелких частиц. При старении свежего осадка в воде или маточном растворе
наряду с изменениями химического состава (гидролизом и дегидратацией)
происходит кристаллизация. При этом крупные агрегаты распадаются, оставляя
россыпь первичных частиц размером 30-60 ангстрем.
1.2 Катализаторы для процессов дегидрирования метанола
При дегидрировании метанола на гетерогенных катализаторах в качестве продуктов чаще рассматриваются метилформиат и формальдегид, хотя известны и более сложные соединения, например диметилметан (метаналь). Все эти вещества широко используются далее при получении полимерных материалов, поэтому вопросы исследования механизма этих реакций и подбора катализаторов достаточно подробно освещены в литературе [2,5]. Метилформиат образуется на медьсодержащих катализаторах в диапазоне температур 200-300 ˚С, где в качестве промоторов используются щелочные металлы, т.к. активность и селективность катализатора зависит от кислотно-основных свойств поверхности. При более высоких температурах реакции, равновесная концентрация метилформиата увеличивается, поскольку реакция эндотермическая, однако, начинает преобладать маршрут его разложения на водород и монооксид углерода [22]. В работах Розовского А.Я. обнаружено, что интенсивность маршрута разложения зависит от парциального давления метанола и метилформиата в реакционной смеси, что принципиально позволяет организовать процесс получения метилформиата не достигая глубокой степени переработки метанола за один проход через реактор [23]. В работе [24], исследованы катализаторы CuO/ZnO с различными соотношениями меди и цинка, в данной реакции и установлено, что метилформиат, наряду с маршрутом дегидрирования метанола может получаться по реакции карбонилирования метанола. Это имеет место на катализаторах с высоким содержанием оксида цинка и большой концентрации монооксида углерода в реакционной смеси.
На медьсодержащих катализаторах наряду с метилформиатом можно получить и формальдегид. В частности, в работе [25] исследовали нанесенную систему CuO/слюда, где состояние меди направленно изменяли от Сu2+ до Cu0, и было установлено, что на не восстановленной меди (Сu2+) при температуре до 240 ˚С образуется только метилформиат, а на металлической меди в интервале температур 240-400 ˚С только формальдегид. В катализаторах Сu0/SiO2 имеет место образования как метилформиата, так и формальдегида.
Авторы работы [26] для подобной системы (Сu0/SiO2) констатировали, что в зависимости от состоянии меди: мелкие кристаллы СuО, кластеры Сu-O-Cu, изолированные ионы меди Cu-O-Si и парциальном давлении метанола в реакционной смеси, наряду с метилформиатом образуется диметоксиметил или происходит глубокое окисление метанола до диоксида углерода.
Методами ИК-спектроскопии показано, что разложение метанола происходит с образованием метокси групп (СН3О-), формильных групп (О-сН2), который близок по структуре хемосорбированному формальдегиду. Эти группировки взаимодействуют с поверхностными гидроксидными группами (ОН) образующие поверхностные форматы (), которые могут разлагаться или на диоксид углерода и водород, или на монооксид углерода и гидроксидную группу. Соотношение между этими группировками и скоростью взаимных переходов в конечном итоге определяют селективность катализатора [27].
В последнее время большой интерес вызывает реакция паровой конверсии метанола с получением водорода, который также проводится на медных катализаторах при температуре порядка 250 ˚С [28,29]. В данной реакции высокое парциальное давление водяного пара в системе ингибирует маршруты образования различных углеводов и продуктами является только водород и диоксид углерода. В частности, в работе [30] показано, что после удаления хемособрбированной воды, которая собственно участвует в реакции паровой конверсии основанной реакцией конверсии метанола, становится образование метилформиата. Для получения формальдегида предложен более широкий круг гетерогенных катализаторов, причем температурный диапазон их работы существенно выше. Для этой цели пригодны катализаторы, содержащие оксиды цинка, титана, натрия, в том числе и нанесенные системы на основе пористого диоксида кремния [31,32]. В тоже время, их селективность и стабильность кажутся недостаточными для промышленного использования. Из последних достижений в этой области следует отметить катализаторы Ag-SiO2-MgO-Al2O3 приготовленных так называемым золь-гель методом, заявленная степень переработки метанола и селективность по формальдегиду достигает 100% [33].
Дегидрирование метанола до формальдегида констатировано, как и на определенных гранях монокристаллов меди, так и на модельных медьсодержащих катализаторах, причем имеет место некоторая конкуренция маршрутов образования формальдегида и метилформиата [34,35].
Разложение адсорбированного метанола на оксиде цинка также сопровождается образованием формальдегида, однако температурный диапазон данной реакции существенно выше, чем на медных системах [36]. Медь - цинковая композиция, является основой низкотемпературных катализаторов, для синтеза метанола и паровой конверсии монооксида углерода. Причем, оксид цинка, наряду с функцией диспергирования обеспечивает возможность обратимых фазовых переходов медного компонента и резервуара активного водорода в каталитическом процессе [37].