Материал: подготовка к экзу фх2

Реакция второго порядка: |
C |
= |
|
|
C0 |
|
C0 |
= |
|
|
|
C0 |
|
|
|
C |
kt |
|
+1=2 t |
|
= |
1 |
|||||||
C0 kt+1 |
2 |
|
C |
0 kt1/ 2+1 |
|
|
kC0 |
||||||||||||||||||||||
|
A |
|
|
|
|
|
|
|
0 |
|
1/ 2 |
|
|
|
1/ 2 |
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
1 |
|
|
|
|
|
|
1 |
1 |
1 |
|
|
|
|
|
3 |
|
|
|
|
|||||||
Реакция третьего порядка: |
C A=√ |
|
|
|
|
k= |
|
( |
|
− |
|
) t1 / 2= |
|
|
|
|
|
||||||||||||
kt+ |
1 |
|
2t |
C 2 |
C02 |
2kC02 |
|
|
|
|
|||||||||||||||||||
|
C02 |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
Степень превращения — это отношение количества молей вещества, вступившего в реакцию,
к исходному числу молей: α |
= |
n |
= |
C0 −C |
=1− |
C |
. |
|
ni |
C0 |
C 0 |
||||||
i |
|
|
|
|
Интегральные методы нахождения порядка реакции.
1)Метод подстановки — подставляем полученные значения времени и концентрации исходного вещества в кинетические уравнения разных порядков, находим константы реакции. Порядок реакции тот, в каком уравнении константа остается постоянной для всех значений.
2)Графический метод — строим графики в координатах C – t, lnC – t, 1/C – t, 1/C2 – t. Какой из этих графиков окажется прямой линией, такой порядок у реакции.
3)Способ определения времени полупревращения — проводим несколько опытов при различных начальных концентрациях и определяем периоды полупревращения. По зависимости периода полупревращения от начальной концентрации определяем порядок реакции. В рамках этого способа можно вычислить порядок реакции по этой
формуле: n= lg t'1'/ 2−lg t'1/ 2 +1 , зная периоды полупревращения при двух различных lg c''0 −lg c'0
начальных концентрациях. Порядок реакции может быть дробным.
4)Метод Эмануэля-Кнорре позволяет вычислить порядок реакции по формуле:
|
ln[ |
t ' |
|
] |
|
|
|
||
n=1+ |
(t ' ' − |
t ') |
, где α '=1− |
c' |
- степень превращения вещества в момент |
||||
lg (1−α ') |
|
c0 |
|||||||
|
|
|
|||||||
времени t', t' – произвольно выбранный момент времени, t'' – такой момент времени, в который c'' = (c')2.
Дифференциальные методы определения порядка реакции.
Реакция aA + bB → продукты.
|
1 dcA |
nA |
nB |
|
|||
Скорость реакции r=− |
|
|
|
=k1 cA |
cB |
, где nA, nB – частные порядки реакций. |
|
a dt |
|||||||
|
|
|
|
||||
Метод изоляции Оствальда. Проводим нужную реакцию сначала в условиях избытка всех исходных веществ, кроме одного. Тогда можно пренебречь изменением концентраций всех реагентов, кроме того, по которому ведется расчет. Тогда концентрацию всех компонентов можно внести в постоянный коэффициент, и скорость реакции можно записать так:
−rA=− |
dc1 |
=k1 c1n1 , где |
k1=akc20n2 |
, n1 – порядок реакции по первому веществу. |
|
||||
|
dt |
|
|
|
Способ логарифмирования |
|
|
||
Прологарифмируем это выражение: |
ln−rA=ln k 1+n1 ln c1 . |
|||
Построим график в координатах ln(-rA) – lnc1, все точки должны лечь на прямую линию.
Тангенс угла наклона этой прямой будет равен порядку реакции по веществу А. Этим методом определяется концентрационный порядок.
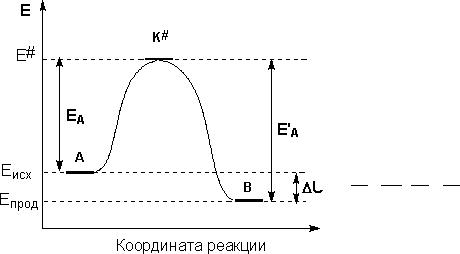
Влияние температуры на скорость химической реакции
Влияние температуры на константу скорости приближенно определяется правилом ВантГоффа: при повышении температуры на каждые 10 градусов скорость элементарной
|
k2 |
T 2−T1 |
|
гомогенной реакции увеличивается в 2-4 раза. |
=γ 10 , где k2 – константа при новой |
||
|
|||
|
k1 |
||
температуре, k1 – константа при исходной температуре, γ — коэффициент Вант-Гоффа. Коэффициент Вант-Гоффа является функцией температуры, при очень высоких и очень низких температурах он становится равным единице, т. е. реакция перестает зависеть от изменения Т.
Более полно зависимость скорости реакции от температуры описывает уравнение Аррениуса:
дlnk |
= |
Ea |
, где Ea – энергия активации — минимальная избыточная энергия по |
|
дT |
RT 2 |
|||
|
|
сравнению со средней, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
|
Ea |
; k= Ae− |
Ea |
|
|
Проинтегрировав это уравнение, получаем: lnk=lnA− |
RT |
. A – |
|||
RT |
|||||
|
|
|
|
предэкспоненциальный множитель — численно равен константе скорости химической реакции при бесконечно большой температуре.
Построив график в координатах lnk – 1/T, получим прямую линию, которая отсекает на вертикальной оси отрезок длиной lnA, а тангенс угла наклона которой равен Ea/R.
Для расчета энергии активации необходимо знать значений констант скоростей при различных температурах, чтобы построить данный график.
Уравнение Аррениуса позволяет рассчитать константу скорости при определенной температуре, зная константу скорости при другой температуре:
ln kk21 = ERa (T11 − T12 )
Для определения энергии активации в первом приближении достаточно знать две константы скорости при двух различных Т.
Температурный коэффициент Вант-Гоффа можно связать с энергией активации:
|
|
k |
2 |
|
|
|
Ea |
( |
1 |
|
|
|
|
1 |
) |
k |
2 |
=exp[ |
Ea |
( |
1 |
|
1 |
)]=γ |
T 2−T1 |
|
|
||||||||||||
ln |
= |
|
− |
− |
10 |
|
|
|
|||||||||||||||||||||||||||||||
k |
1 |
R |
T 1 |
T 2 |
k |
1 |
R |
T 1 |
T 2 |
|
|
||||||||||||||||||||||||||||
|
|
E T −T |
|
|
10 |
|
|
|
|
|
|
|
E T −T |
|
10 |
|
|
|
E |
10 |
|
||||||||||||||||||
|
|
|
T |
−T |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
γ =exp[ |
|
a |
|
2 1 |
] |
2 |
|
|
|
1 =exp[ |
|
a |
|
2 1 |
|
]=exp[ |
a |
|
] |
||||||||||||||||||||
|
R |
T 1 T 2 |
|
|
|
|
|
R |
T 1 T 2 |
T 2−T 1 |
R |
T 1 T 2 |
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
lnγ = |
|
10 Ea |
Ea =0,1 R T 1 T 2 ln γ |
|
|
|
|
|
||||||||||||||||||||||||
|
|
|
|
|
|
|
RT 1 T 2 |
|
|
|
|
|
|||||||||||||||||||||||||||
Энергий активации, которая находится из уравнения Аррениуса для сложных реакций является функций энергий активации элементарных реакций, составляющих сложную реакцию. Эта энергия называется эффективной энергией активации.
Кинетика сложных реакций
При рассмотрении кинетики сложных реакций делается допущение о независимом протекании элементарных стадий, т. е. что величина константы скорости элементарной химической реакции не зависит от того, протекают ли в данной системе одновременно другие элементарные реакции.
Обратимые реакции первого порядка
Обратимые элементарные реакции первого порядка состоят из прямой и обратной элементарных реакций первого порядка: A <=> B. k1 – константа скорости прямой реакции, k2
– обратной.
Скорость обратимой реакции равна разности скоростей прямой и обратной реакций:
w=w1−w2=k1 cA−k 2 cB , где cA, cB – концентрации веществ A и B в момент времени t.
Пусть cA = a – x, cB = b+ x, где x — убыль вещества A за время t с момента начала реакции, a, b - начальные концентрации веществ A и B. Тогда:
dxdt =k 1 (a− x)−k 2 (b+x)=(k1 +k2 )( k1 a−k2 b −x)
При наступлении равновесия скорости прямой и обратной реакции равны, а концентрация веществ не изменяется:
dx |
=0=(k1+k 2)( |
k1 a−k2 b |
− x) x= |
k1 a−k 2 b |
=x∞ |
, где x∞ - равновесная убыль |
|
dt |
k1+k 2 |
k 1+k2 |
|||||
|
|
|
|
концентрации, т. е. то изменение концентрации, которое произойдет от начала реакции до наступления равновесия. Тогда перепишем уравнение:
dxdt =(k1 +k2 )( x∞− x)
k |
+k |
= |
1 ln |
x∞ |
|
x∞− x |
|||||
1 |
2 |
|
t |
k1 > k2 |
k1 = k2 |

k1 < k2
Параллельные реакции первого порядка
К параллельным реакциям относятся химические превращения, когда взятое для реакции вещество претерпевает одновременное изменение в двух и более направлениях:
A →k1→ B, A →k2→ D.
Скорость расходования вещества А находится как сумма скоростей обеих реакций с учетом правила знаков:
w=−w1−w2=− |
dcA |
|
=k1 cA+k2 cA=(k1+k2 )cA |
|
, a – начальная концентрация вещества А. |
||||||||||||||||||||||
dt |
|
|
|||||||||||||||||||||||||
|
|
cA=a e−(k1+k 2)t |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Найдем концентрации веществ B и D: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
dc B |
|
|
|
−(k |
1+ k2)t |
|
|
|
|
|
|
k1 a |
|
|
−(k1+k 2)t |
|
|
|
|
|
|
|||||
|
|
=k1 cA dc B=k |
1 a e |
|
dt cB= |
|
|
|
|
|
|
(1−e |
|
|
) |
|
|
|
|
||||||||
|
dt |
|
|
k1+k2 |
|
|
|
|
|||||||||||||||||||
|
dcD |
|
|
|
−(k |
1+k2)t |
|
|
|
|
|
|
k 2 a |
|
|
|
−(k 1+k2)t |
|
|
|
|
|
|
||||
|
dt |
=k2 cA dc B=k |
2 ae |
|
dt cD= |
|
|
|
|
(1−e |
|
|
) |
|
|
|
|
||||||||||
|
|
k1+k 2 |
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
Для нахождения констант скоростей каждой реакции можно |
||||||||||||||||||||
|
|
|
|
|
|
|
воспользоваться следующей системой уравнений: |
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
−(k1+ k2)t |
|
|
|
|
|
|
1 |
a |
|||||||||
|
|
|
|
|
|
|
c A=a e |
|
|
|
|
ln cA=ln a−(k 1+k2 )t k1+k 2= t ln |
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
cA |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
cB |
= |
k 1 |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
cD |
k 2 |
|
|
|
||
|
|
|
|
|
|
|
Итак, зная концентрации всех веществ в текущий момент |
||||||||||||||||||||
|
|
|
|
|
|
|
времени, а также начальную концентрацию исходного |
||||||||||||||||||||
|
|
|
|
|
|
|
вещества, константы скоростей каждой из реакций находятся |
||||||||||||||||||||
|
|
|
|
|
|
|
путем решения системы уравнений: |
|
|
||||||||||||||||||
|
|
k1 > k2 |
|
|
|
|
|
k1 |
= |
cB |
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
k2 |
cD |
|
|
a |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
k |
+k |
|
|
|
= 1 ln |
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
cA |
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
{1 |
|
|
2 |
|
t |
|
|
|
|
|
|
|
|
|
||||||
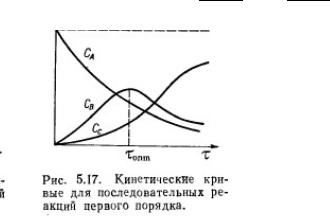
Последовательные реакции первого порядка
В последовательных реакциях промежуточные вещества, которые образуются в одной стадии, расходуются в последующей:
A → k1 → B → k2 → D
Обозначим концентрации веществ в различные промежутки времени: t = 0: cA = a, cB = 0, cD = 0
t:cA = a-x, cB = x-y, cD = y
t = t∞: cA = 0, cB = 0, cD = a
x – убыль вещества A, y – увеличение количества вещества D. Вещество А расходуется на реакцию k1:
−d (a−x ) |
=k1 |
(a− x) d (a− x) |
=−k 1 dt a−x=ae−k1 t |
dt |
|
a− x |
|
Вещества B образуется в результате реакции k1 и расходуется на реакцию k2:
|
d ( x− y ) |
|
|
|
k 1 a |
−k1 t |
−k2 t |
|
|
− |
|
=−k1 |
(a−x)+k2 |
( x− y) x− y= |
|
(e |
−e |
) |
|
dt |
k 2−k1 |
||||||||
|
|
|
|
|
|
|
Вещество D образуется в результате реакции k2:
−dydt =−k 2 ( x− y) y=a (1− k 2k−2k 2 e−k1 t + k2k−1k 2 e−k2 t )
Время от начала реакции, когда достигается максимум концентрации промежуточного вещества, можно рассчитать, зная, что производная в этой точке равна 0:
|
|
|
|
|
|
|
ln |
k 2 |
|
|
||
− |
d ( x− y ) |
=0 t |
|
|
= |
k 1 |
|
|||||
|
|
|
|
|||||||||
dt |
max |
k 2−k 1 . |
||||||||||
|
|
|
|
|||||||||
|
( x− y )max=a(kk |
12 ) |
|
k2 |
||||||||
|
k1−k2 |
|
||||||||||
В это же время на кинетической кривой продукта
реакции наблюдается перегиб.
Период индукции — время от начала реакции до того момента, когда становится заметно отличие от нуля скорости реакции. Период индукции можно рассчитать, проведя касательную к кривой вещества D в точке перегиба и найти ее пересечение с осью времени.
Чем больше отношение k2 к k1, тем левее и ниже будет находиться максимум концентрации промежуточного вещества.
Метод квазистационарных концентраций
Метод применяется в том случае, когда в ходе реакции образуются неустойчивые промежуточные вещества. Если скорость распада этих веществ намного превышает скорость их образования, то их концентрация в любой момент времени очень мала. В этом случае скорость образования этих веществ можно принять за ноль. Этот метод позволяет выражать концентрацию промежуточных веществ через концентрации исходных веществ, что значительно упрощает анализ сложных реакций.
Стационарный режим протекания реакции может установиться в открытой системе. При