Материал: Катализатор каталитического крекинга и каталитического риформинга
В связи с возросшими требованиями к экологической безопасности промышленных процессов исключительно актуальной становится проблема улавливания вредных компонентов газовых выбросов.
Если в состав ЦСК ввести твердую добавку MgO или СаО, то такой катализатор становится переносчиком оксидов серы из регенератора в реактор по схеме:
в регенераторе: MgO + SO3 → MgSO4 ;
в реакторе: MgSO4 + 4Н2 → MgO + H2S + 3H2O ;
или 2MgSO4 + СН4 → 2MgO + 2H2S + СО2 .
Образующийся сероводород, выводимый из реактора вместе с продуктами крекинга, будет извлекаться затем из газов аминной очисткой;
д) для повышения механической прочности ЦСК в состав аморфной матрицы дополнительно вводят тонкодисперсную окись алюминия (α-форму). Кроме того, для снижения потерь катализатора от испарения и уменьшения коррозии аппаратуры в системах катализатора в циркулирующий катализатор вводят смазывающие порошки из смеси окиси магния, карбоната и фосфата кальция, иногда титаната бария. Эти добавки взаимодействуют при высокой температуре с поверхностью катализатора, в результате чего на ней образуется глянец, способствующий снижению истирания.
2.1.2 Механизм процессов на катализаторе
Химические превращения углеводородов крекируемого сырья, протекающие по карбений-ионному цепному механизму на поверхности ЦСК, можно представить в целом в следующей последовательности.
1. Первичные мономолекулярные реакции крекинга и деалкилирования (распад по С–С-связи) высокомолекулярных молекул исходного сырья с образованием низкомолекулярных (н. м.) углеводородов:
а) крекинг парафинов с образованием н. м. парафина и олефина:
СnH2n+2 → CmH2m + CpH2p+2 ;
б) крекинг олефинов с образованием н. м. олефинов:
СnH2n → CmH2m + CpH2p ;
в) деалкилирование алкилароматических углеводородов:
ArCnH2n+1 → ArH + CnH2n → ArCmH2m–1 + CpH2p ;
г) крекинг нафтенов с образованием олефинов:
ц-СпН2n → CmH2m + CpH2p ,
где n = m + р.
Первичные реакции распада могут осуществляться либо термически по радикально-цепному механизму, либо каталитически на апротонных (льюисовских) центрах алюмосиликатной матрицы ЦСК:
RH + L → R+ + LH–
R+ → н. м. олефин + R+'
R+' + LH → R'H + L или
R+' → H+ + олефин
2. Вторичные бимолекулярные реакции углеводородов на поверхности цеолита с участием карбений-ионов, образующихся преимущественно присоединением протона к олефину (инициирование цепи):
RCH = CH2 + HA → RCHCH3 + A—
Различие по реакционной способности образующихся карбкатионов обусловливает вероятные направления превращений и степень участия их в дальнейших реакциях. Установлено, что стабильность карбениевых ионов возрастает в ряду:
СН3 < +С2Н5 < + первичный < вторичный < третичный.
Третичный карбениевый ион является самым стабильным. Именно этим обусловлен высокий выход изопарафиновых углеводородов, особенно изобутана, при каталитическом крекинге.
Реакции развития цепи включают следующие наиболее характерные реакции карбениевых ионов: распад С–С-связи, перенос гидридиона (Н-перенос), изомеризацию, циклизацию, дециклизацию, деалкилирование, алкилирование, полимеризацию, поликонденсацию и др.
Обрыв цепи превращений карбениевых ионов происходит возвратом протона к поверхности катализатора или отнятием электрона от центров Льюиса.
Распад С–С-связи карбений-иона является одной из наиболее важных целевых реакций, приводящих к образованию низкомолекулярных крекинга. Для этой реакции применимы следующие правила:
а) легче всего разрывается С–С-связь, находящаяся в β-положении по отношению к атому углерода, несущему заряд (правило — β-распада);
б) у образующихся олефинов имеется двойная связь у первого углеродного атома;
в) из нескольких возможных вариантов более вероятен β-распад карбений-иона с образованием олефина с меньшей длиной цепи:
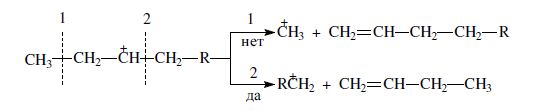
Продукт первичного β -распада — карбений-ион может снова крекироваться до образования более стабильных карбкатионов или углеводородов (после отдачи протона или присоединения электрона);
г) более выгодным для алкилароматических или алкилнафтеновых углеводородов является отрыв всей алкильной группы:
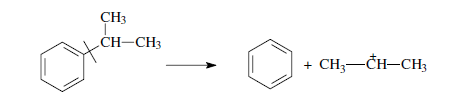
Поскольку образование требует высоких энергетических затрат, цепной распад карбкатионов прерывается до образования карбениевых ионов с числом углеродных атомов 3…5.
Перенос гидрид-иона (Н-перенос) можно проиллюстрировать следующим образом:
![]()
Установлено, что лучшими гидридными донорами являются нафтены, полициклические нафтены или нафтено-ароматические углеводороды, изоалканы и даже олефины. Энергетически более выгоден отрыв гидрид-иона от третичного, затем вторичного и менее выгоден от первичного углеродного атома. Нафтеновые, алкилароматические и изопарафиновые углеводороды часто содержат третичные атомы углерода и поэтому интенсивно участвуют в реакциях Н-переноса. Активными акцепторами гидрид-ионов являются наименее стабильные высокореакционноспособные карбений-ионы или углеводороды, содержащие несколько π-связей, например диолефины. Именно Н-перенос обусловливает повышенные выход топливных фракций и химическую стабильность бензинов каталитического крекинга. По Н-переносу осуществляются следующие реакции каталитического крекинга:
олефин + нафтен парафин + арен
олефин + парафин парафин + олефин
олефин + олефин арен + парафин
олефин + олефин арен + водород
арен + арен кокс + парафин + водород и т. д.
Изомеризация карбениевых ионов является наряду с распадом важной целевой реакцией, повышающей товарные качества продуктов каталитического крекинга.
В большинстве случаев изомеризация протекает быстрее, чем крекинг, и потому часто предшествует β-распаду. Сочетание реакций изомеризации и β-распада обусловливает повышенное содержание в продуктах каталитического крекинга углеводородов изостроения.
Изомеризация карбениевых ионов может происходить либо путем передачи протона (гидридный сдвиг), либо метильной группы (скелетная изомеризация) вдоль углеводородной цепи:
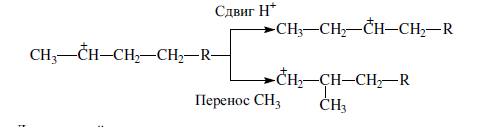
Для реакций изомеризации предложен механизм, согласно которому процесс осуществляется через образование промежуточных циклических структур, например циклопропана, циклобутана и т. д. (по-видимому, посредством многоточечной, т. е. мультиплетной хемосорбции):
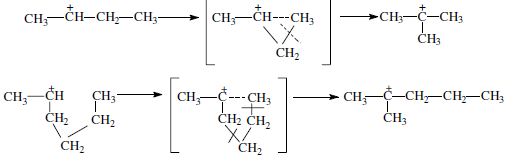
и переносом метильной группы внутри молекулы при изомеризации ди- и полиметилбензолов.
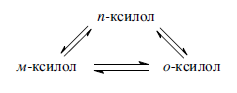
Так, ксилолы подвергаются взаимопревращению:
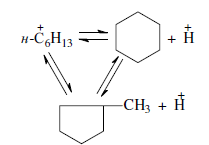
Циклизация и дециклизация как обратимые реакции с участием карбений-ионов протекают, по-видимому, через мультиплетную хемосорбцию:
или через диеновый синтез:
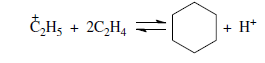
Циклопентаны в условиях каталитического крекинга более устойчивы, чем циклогексаны. Циклогексаны в этих условиях могут подвергаться дегидрированию в арены посредством Н-переноса. При наличии длинных боковых цепей в циклоалкановом карбениевом ионе возможны изомеризация боковой цепи и деалкилирование. Бициклические циклоалкановые карбениевые ионы ароматизируются в большей степени, чем моноциклические.
Алкилирование и полимеризация — реакции, противоположные крекингу, протекают по карбений-ионному механизму. При температурах ниже 400 °С они доминируют над крекингом, а при высоких температурах равновесие смещается в сторону деалкилирования и деполимеризации.
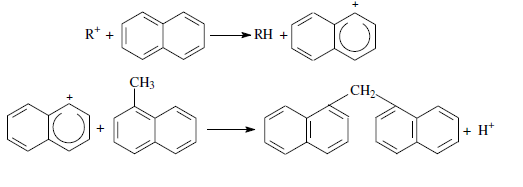
Конденсация ароматических углеводородов, дающая соединения с более высокой молекулярной массой, вплоть до кокса, характерна для каталитического крекинга. При этом ареновый карбений-ион вступает в последовательные реакции присоединения (конденсации) к ароматическим углеводородам и Н-переноса. Процесс конденсации вследствие высокой стабильности полициклического аренового карбений-иона может продолжаться до обрыва цепи:
Коксообразование. При осуществлении реакций углеводородов на кислотных катализаторах образуется углеродистый материал, называемый коксом, который не десорбируется с поверхности катализатора.
Этот материал имеет атомное отношение водорода к углероду от 0,3 до 1,0 и спектроскопические характеристики, аналогичные таковым для полициклических ароматических соединений.
При крекинге ароматических углеводородов кокс получается более обогащенным углеродом, чем при крекинге парафинистого сырья.
В составе кокса крекинга сернистого нефтяного сырья всегда содержится сера. В среднем отношение содержания серы в коксе к ее содержанию в сырье крекинга близко к единице.
Вследствие экранизации активных центров ЦСК коксовыми отложениями активность катализатора крекинга быстро снижается. Эта дезактивация является обратимой, так как после окислительной регенерации первоначальная активность практически полностью восстанавливается.
2.1.3 Промышленный марки катализаторов каталитического крекинга
На отечественных установках с движущимся слоем шарикового катализатора применялись и продолжают пока применяться шариковые катализаторы АШНЦ-3 (без РЗЭ), АШНЦ-6, Цеокар-2 и Цеокар-4 (все с РЗЭ).
Из микросферических ЦСК применение находят: КМЦР-2 (2 % La2O3), МЦ-5 и РСГ-6Ц (по 4 % La2O3), КМЦР-4 (с промотором дожига) и др. Из зарубежных ЦСК более известны следующие марки катализаторов: Дюрабед (5, 6, 8, 9), Супер (Д, экстра Д), (1–7), CBZ (1–4),Октакэт-11, Резидкэт (20, 30) и другие.
2.2 Катализаторы каталитического риформинга
2.2.1 Состав катализаторов каталитического риформинга и механизм
Процесс каталитического риформинга осуществляют на бифункциональных катализаторах, сочетающих кислотную и гидрирующую-дегидрирующую функции. Гомолитические реакции гидрирования и дегидрирования протекают на металлических центрах платины или платины, промотированной добавками рения, иридия, олова, галлия, германия и др., тонко диспергированных на носителе.
Кислотную функцию в промышленных катализаторах риформинга выполняет носитель, в качестве которого используют оксид алюминия.
Для усиления и регулирования кислотной функции носителя в состав катализатора вводят галоген: фтор или хлор. В настоящее время применяют только хлорсодержащие катализаторы. Содержание хлора составляет от 0,4-0,5 до 2,0 % мас.
Бифункциональный механизм доказан на примере использования катализаторов, содержащих только кислотные центры или только металлические центры, которые оказались исключительно малоактивными, в то время как даже механическая их смесь была достаточно активна. Благодаря бифункциональному катализу удается коренным образом преобразовать углеводородный состав исходного бензина и повысить его октановую характеристику на 40...50 пунктов.
Платина на катализаторе риформинга не только ускоряет реакции гидрирования-дегидрирования, но и замедляет образование кокса на его поверхности. Обусловливается это тем, что адсорбированный на платине водород сначала диссоциируется, затем активный (атомарный) водород диффундирует на поверхности катализатора к кислотным центрам, ответственным за образование коксовых отложений. Коксогены гидрируются и десорбируются с поверхности. В этой связи скорость образования кокса при прочих равных условиях симбатно зависит от давления водорода. Поэтому минимальная концентрация платины в катализаторах риформинга определяется необходимостью прежде всего поддерживать их поверхность в «чистом» виде, а не только с целью образования достаточного числа активных металлических центров на поверхности носителя.
В монометаллических алюмоплатиновых катализаторах риформинга содержание платины составляет 0,3…0,8 % мас. Очень важно, чтобы платина была достаточно хорошо диспергирована на поверхности носителя. С увеличением дисперсности платины повышается активность катализатора.
Прогресс каталитического риформинга в последние годы был связан с разработкой и применением сначала биметаллических и затем полиметаллических катализаторов, обладающих повышенной активностью, селективностью и стабильностью.
Используемые для промотирования металлы можно разделить на две группы. К первой из них принадлежат металлы 8-го ряда: рений и иридий, известные как катализаторы гидродегидрогенизации и гидрогенолиза. К другой группе модификаторов относят металлы, практически неактивные в реакциях риформинга, такие как германий, олово и свинец (IV группа), галлий, индий и редкоземельные элементы (III группа) и кадмий (из II группы).
К биметаллическим катализаторам относят платино-рениевые и платино-иридиевые, содержащие 0,3…0,4 % мас. платины и примерно столько же Re и Ir. Рений или иридий образуют с платиной биметаллический сплав, точнее кластер, типа Pt-Re-Re-Pt-, который препятствует рекристаллизации — укрупнению кристаллов платины при длительной эксплуатации процесса. Биметаллические кластерные катализаторы (получаемые обычно нанесением металлов, обладающих каталитической активностью, особенно благородных, на носитель с высокоразвитой поверхностью) характеризуются, кроме высокой термостойкости, еще одним важным достоинством — повышенной активностью по отношению к диссоциации молекулярного водорода и миграции атомарного водорода (спилловеру). В результате отложение кокса происходит на более удаленных от металлических центров катализатора, что способствует сохранению активности при высокой его закоксованности (до 20 % маc. кокса на катализаторе). Из биметаллических катализаторов платино-иридиевый превосходит по стабильности и активности в реакциях дегидроциклизации парафинов не только монометаллический, но и платино-рениевый катализатор. Применение биметаллических катализаторов позволило снизить давление риформинга (от 3,5 до 2...1,5 МПа) и увеличить выход бензина с октановым числом по исследовательскому методу до 95 пунктов примерно на 6 %.