Материал: физхимия метода катализ
3. Электронная теория (с.З. Рогинский)
(по желанию, взяла это с учебника)
В основе теории лежит представление о том, что катализатор имеет свободные или слабосвязанные электроны.
Такими электронами обеспечиваются свободные валентности на поверхности катализатора, за счет которых адсорбируются молекулы реагирующих в-в с образование свободных атомов и радикалов.
При взаимодействии свободных атомов и радикалов образуются продукты реакции.
Например:
Затруднено осуществление реакции H2 + O2 = H2O из-за насыщенности связей реагирующих в-в
Но на платиновом катализаторе свободные электроны переходят к молекуле кислорода, образуя ионы кислорода (2O-)
На положительно заряженной платине адсорбируются молекула водорода, отдавая ей электроны и превращается в катионы (2H+)
Ионы водорода и кислорода взаимодействуют между собой и образуют воду
Кислотно-основный катализ
Кислотно-основный катализ – ускорение реакций кислотами и основаниями.
Такая способность кислот и оснований – один из признаков характеризующих этот класс химических соединений.
Классическая теория кислотно-основного катализа приписывала каталитическое действие исключительно иона водорода и гидроксила. Позднее было установлено, что реакции катализируются также недиссоциированными молекулами кислот и оснований и различными другими ионами, например ионом NH4+.
Теория общего кислотно-основного катализа является составной частью теории кислот и оснований Бренстеда-Лоури:
Кислота – донор протона
Основание – акцептор протона
Кислотами и основаниями могут быть электронейтральные молекулы, катионы и анионы с различным зарядом.
Кислотно-основное взаимодействие в средах с высокой диэлектричской проницаемостью состоит в передаче протона от кислоты HA к основанию B с образованием новой пары – кислоты BH+ и основания A- :
HA + B ↔ BH+ + A⁻
Кислотно-основный катализ обусловлен протолитической реакцией между субстратом, выступающим в роли слабой кислоты или слабого основания, и катализатором, которым могут быть соединения, удовлетворяющие определению кислот и оснований по Бренстеду.
Протолитическая реакция (присоединение или отщепление протона), как стадия каталитичского процесса, приводит к перераспределению электронов в молекуле субстрата и тем самым к образованию промежуточных соединений с повышенной реакционной способностью (карбкатионы, полярные комплексы). Эта стадия реакции обусловливает снижение энергии активации и ускорение процесса в целом.
С излагаемой точки зрения кислотно-основный катализ обязательно включает стадию переноса протона из одного места молекулы в другое и поэтому требует присутствия как доноров, так и акцепторов протонов, что отвечает следующей схеме:
Кислотный катализ |
Основный катализ |
HX + HA ⇄ HXH+ + A⁻ HXH+ + B ⇄ XH + BH+ A⁻ + BH+ ⇄ HA + B |
HX + B ⇄ BH+ + X⁻ X- + HA ⇄ XH + A⁻ A⁻ + BH+ ⇄ HA + B |
Где HX – субстрат; XH – продукт реакции; HA – кислота; B – основание
Пример кислотного катализа: кето-енольная таутомерия, определяющая протекание реакции йодирования ацетона.
В результате протолитической реакции кетон переходит в енольную форму:
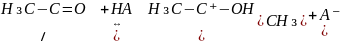
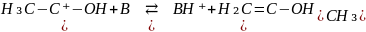
Енольная форма легко реагирует с йодом:
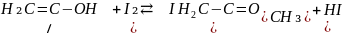
Пример основного катализа: можно проиллюстрировать реакцией альдольной конденсации альдегидов и кетонов:
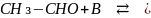 ⁻
⁻
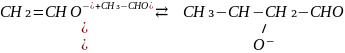
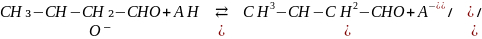
HA – кислота;
A⁻ - основание, сопряженное с кислотой HA;
B – основание;
BH⁺ - кислота, сопряженная с основанием B.
Конкретный пример кислотного катализа: получение сложного эфира
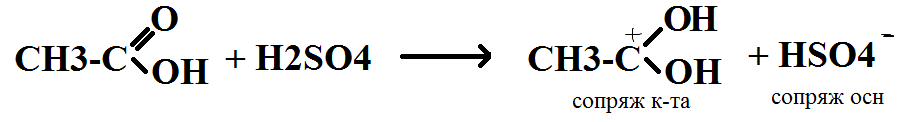
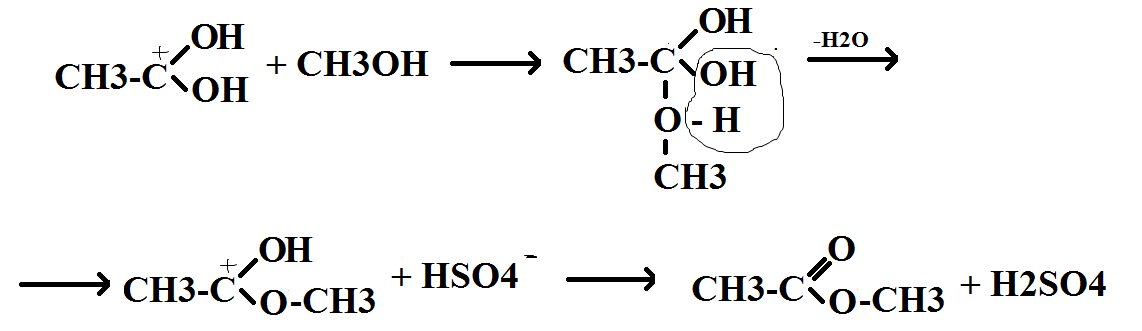
Реакции кислотно-основного катализа протекают не только в растворах, но также на поверхности твердых катализаторов, обладающих свойствами кислот и оснований
Перенос протона от кислоты к основанию, является частным случаем донорно-акцепторного взаимодействия общего типа, т.е. образования координационной связи между акцепторами и донорами электронных пар и гетеролитического расщепления таких связей.
Координационно-комплексный катализ
Сущность каталит. действия заключается в том, что ионы металлов выступают как доноры или акцепторы электронов
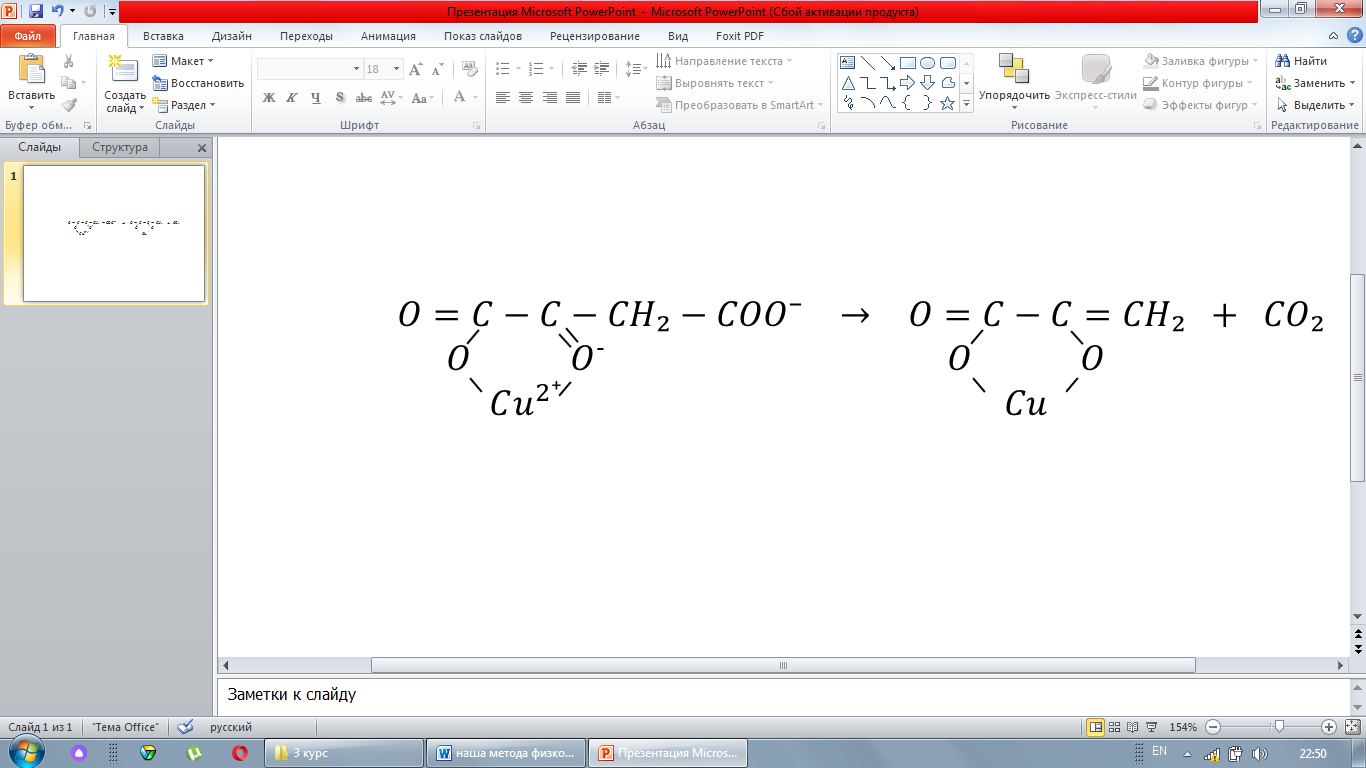
Пример 1: реакция декарбоксилирования щавелево-уксусной кислоты может ускориться ионом H⁺ или ионом металла, связывающем кислоту в комплекс:
Пример 2: реакция Кучерова
Ионы Н+ уменьшают отрицательный заряд и увеличивают легкость акцептирования остающейся молекулой пары электронов из отщепляющейся карбоксильной группы.
Однако еще лучше эту функцию выполнит многозарядный ион металла, связывающий анион дикетокислоты в прочный комплекс, как указано на схеме.
Для реакции декарбоксилирования найден порядок каталитической активности ионов: примерно соответствующий их способности к комплексообразованию.
Al 3+ > Fe 3+ > Cu 2+ >Fe 2+ > Zn 2+ > Mg 2+ > Mn2+ > Ca2+
В общем, ионы металлов будут катализировать реакции, ускоряемые кислотами, если при этом образуются промежуточные хелатные соединения.
!!! Каталитические свойства иона металла обусловлены его поляризующим действием, то есть отношением квадрата его заряда к радиусу (e2/r).
За счет поляризации снижается активационный барьер реакции.
Иногда бывает, что хорошие комплексообразователи с большим (e2/r) каталитически мало активны. Это относится к переходным металлам.
Для объяснения этого отклонения выдвинуто представление о том влиянии, которое оказывают изменение координации лигандов вокруг комплексного иона переходного элемента на его реакционную способность. Согласно этим взглядам, для каталитической реакции может быть существенна конфигурация орбит d-электронов иона-катализатора. В поле лигандов происходит расщепление d-уровней этого иона на подуровни, причем электроны стремятся занять низкие d-подуровни. Напротив, изменение координации вокруг переходных элементов, а также ионов со структурой d0 (Sc3+, Ti4+) и d5 (Fe3+, Mn2+ ) не вызывает изменений электронной конфигурации. Поэтому каталитические свойства ионов переходных и непереходных металлов должны отличаться. Кроме того, важное значение имеет просто геометрический размер иона. Известно, что ион Be2+ не способен катализировать бромирование ацетоуксусного эфира, так как кроме выше сказанного, он еще и просто мал, для того, чтобы разместить на себе два лиганда в активированном комплексе.
В живых организмах активаторами биокаитализаторов – ферментов также служат ионы- комплексооборазователей: марганец (2+), железо (2+; 3+), кобальт (2+), никель (2+), медь (2+), магний (2+). Комплексы этих ионов с органическими молекулами катализируют многие окислительно-восстановительные реакции и реакции гидролиза. Нпример, Mg2+ входит в состав хлорофилла, а Fe2+ в состав гемоглобина, а Fe3+ - в состав каталазы. В ряде случаев была обнаружена зависимость каталитической активности этих ионов от природы лиганда. Особенно сильно активируют азотосодержащие лиганды.
Увеличение константы скорости «k» при катализе комплексами одного и того же иона с различными лигандами, происходит засчет множителя k0 в уравнении Арениуса. В реакциях этого типа часто наблюдается постоянство энергии активации.
Ферментативный катализ
Подавляющее большинство реакций, протекающих в организмах, осуществляется при помощи биологических катализаторов, которые имеют общее название "ферменты".
Ферменты – белки, входящие в состав клеток и тканей, катализирующие хим. реакции, протекающие в организме.
Характерная особенность ферментов - их специфичность
(свойство изменять скорость реакции одного типа и не влиять на многие другие реакции, протекающие в клетке)
Специфичность ферментов подразделяется на:
Абсолютная специфичность (химическая) – действие каждого фермента на вещества строго определенного состава. Например, уреаза катализирует гидролиз мочевины, пепсин - расщепление белков, каталаза действует только на пероксид водорода.
Стереохимическая специфичность заключается в том, что ферменты действуют только на определенные стереоизомеры органических соединений.
Причины высокой специфичности:
Теория Фишера "замок-ключ" – молекула субстрата (ключ) точно соответствует по своей форме некоторому участку на молекуле фермента. При этом не происходит нарушения формы обеих молекул.
Теория Кошланда (рука-перчатка) – он несколько видоизменил модель "ключ-замок". Согласно его гипотезе, субстрат присоединяется к активному центру, изменяет его форму, обеспечивая таким образом идеальное их соответствие.
Образование Ф-S комплекса может происходить за счет электрически заряженных группировок как на ферменте, так и на субстрате.
Такими группировками могут быть RCOO- или R-NH3+.
В результате такого взаимодействия в субстрате могут происходить определенные химические изменения, выражающиеся в образовании новых функциональных групп с совершенно иными полярными свойствами.
Т.о. специфичность фермента обусловлена его конфигурацией, строением и электрическими свойствами активной группы фермента.
Инактивация – явление разрушения фермента и утраты его активности в процессе протекания каталитической реакции.
Чем активнее фермент, тем он сильнее разрушается и значительная чувствительность к изменению внешних условий, температуры и рН среды.
Влияние рН на активность фермента объясняется изменением состояния ионизации не только фермента и субстрата в отдельности, но и фермент-субстратного комплекса.
Ферментативная реакция в целом протекает по схеме:
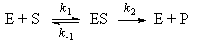
E - фермент, S - субстрат
ES - субстрат-ферментный комплекс,
P - продукт реакции
k1, k-1, k2 - константы скоростей W1, W-1 и W2.
Наиболее медленной стадией является распад фермент-субстратного комплекса на продукты реакции и фермент. Значит, эта стадия будет определять скорость ферментативной реакции
W2=K2[ES];
Эта скорость достигает max значения, когда весь фермент [E0] окажется связанным в виде фермент-субстратного комплекса:
Wmax=k2[E0]
Согласно закону действующих масс
- скорость образования субстрат-ферментного комплекса будет равна:
W1=k1[S][E]
- скорость распада субстрат-ферментного комплекса W-1 будет равна:
W-1=k-1[SE].
При достижении стационарного состояния скорость образования субстрат-ферментного комплекса W1 будет равна скорости его распада по обоим направлениям:
W1=W-1+W2
k1[S]{[E0+]-[ES]}=k-1[ES]+k2[ES].
Преобразуем:
k1[S][E0]=k1[S][ES]+k-1[ES]+k2[ES]
или
k1[S][E0]=[ES]{k1[S]+(k-1+k2)};
Разделим на k1
[S][E0]=[ES]{[S]+ (k-1+k2)/k1}.
(k-1+k2)/k1=Km
Выразим [ES]:
[ES]=[S][E0]/([S]+Km)
Т.к за образование продуктов р. отвечает W2, то для общей скорости реакции:
W2=k2[ES]=k2[S][E0]/(Km+[S])
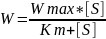
ур-е Михаэлеса-Мента
где Wmax = [E0]* k2 – это максимальная скорость, когда весь катализатор связан
При скорости ферментативной реакции
W2=1/2max
Km=[S].
Km – константа Михаэлеса
График зависимости скорости ферментативной реакции от концентрации субстрата выглядит следующим образом:
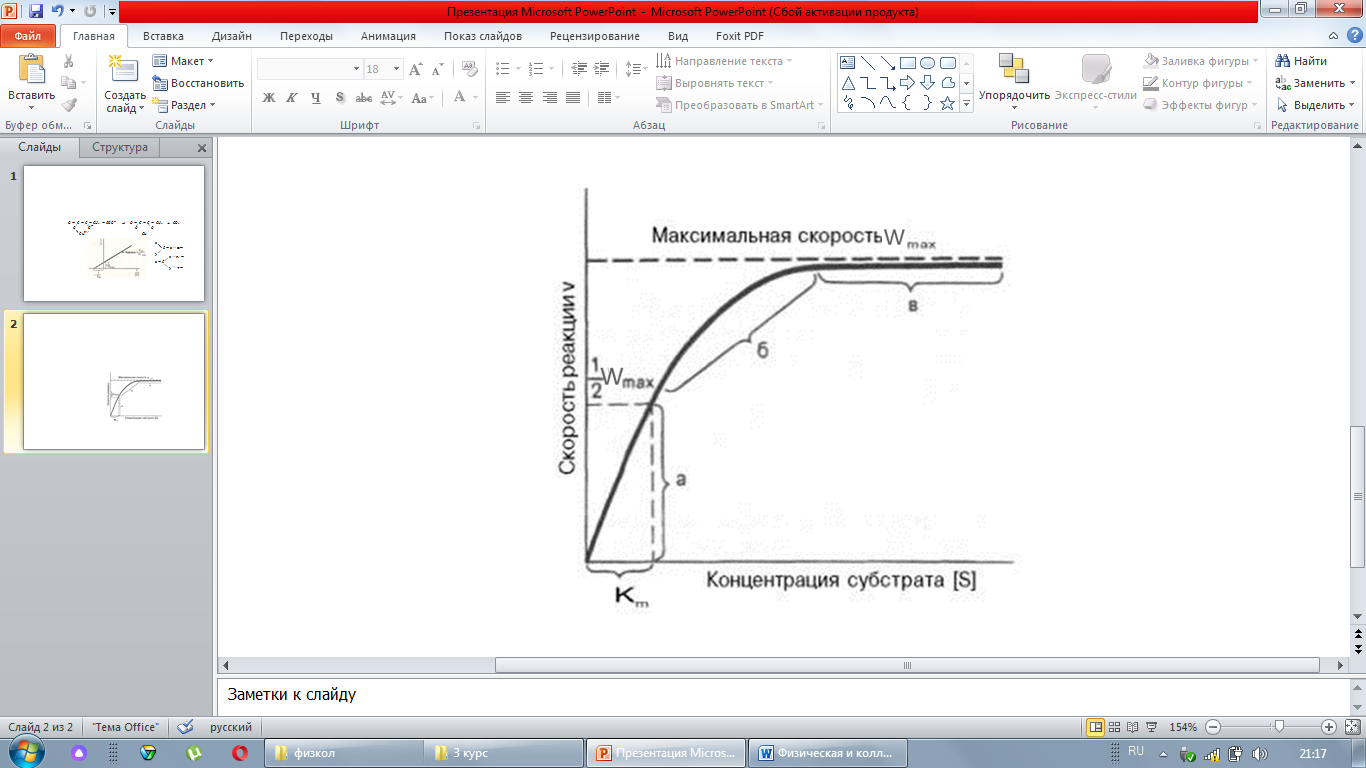
При небольших концентрациях [S] молекулы субстрата полностью располагаются на активных центрах и скорость ферментативной реакции прямо пропорциональна этой концентрации. Это участок ОА, отвечающий реакции 1-ого порядка. По мере занятости активных центров скорость падает и достигает постоянной величины.
В классическом виде уравнение Михаэлиса-Ментен выглядит следующим образом:
W=Wmax*[S]/(KS+[S])
Km=(k-1+k2)/k+1=KS+k2/k+1
k-1/k+1=KS – константа диссоциации субстрат-ферментного комплекса или субстратная константа.
KS зависит от природы субстрата и фермента и определяет степень их сродства. Чем меньше KS, тем больше их сродство.
Уравнение: W=Wmax*[S]/(Km+[S]) называется уравнением Бригса-Холдейна
Для более удобного графического представления экспериментальных данных Берк и Лайнуивер преобразовали уравнение Бригса-Холдейна, исходя из принципа: если существует равенство между двумя величинами, то равны и их обратные величины;
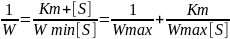
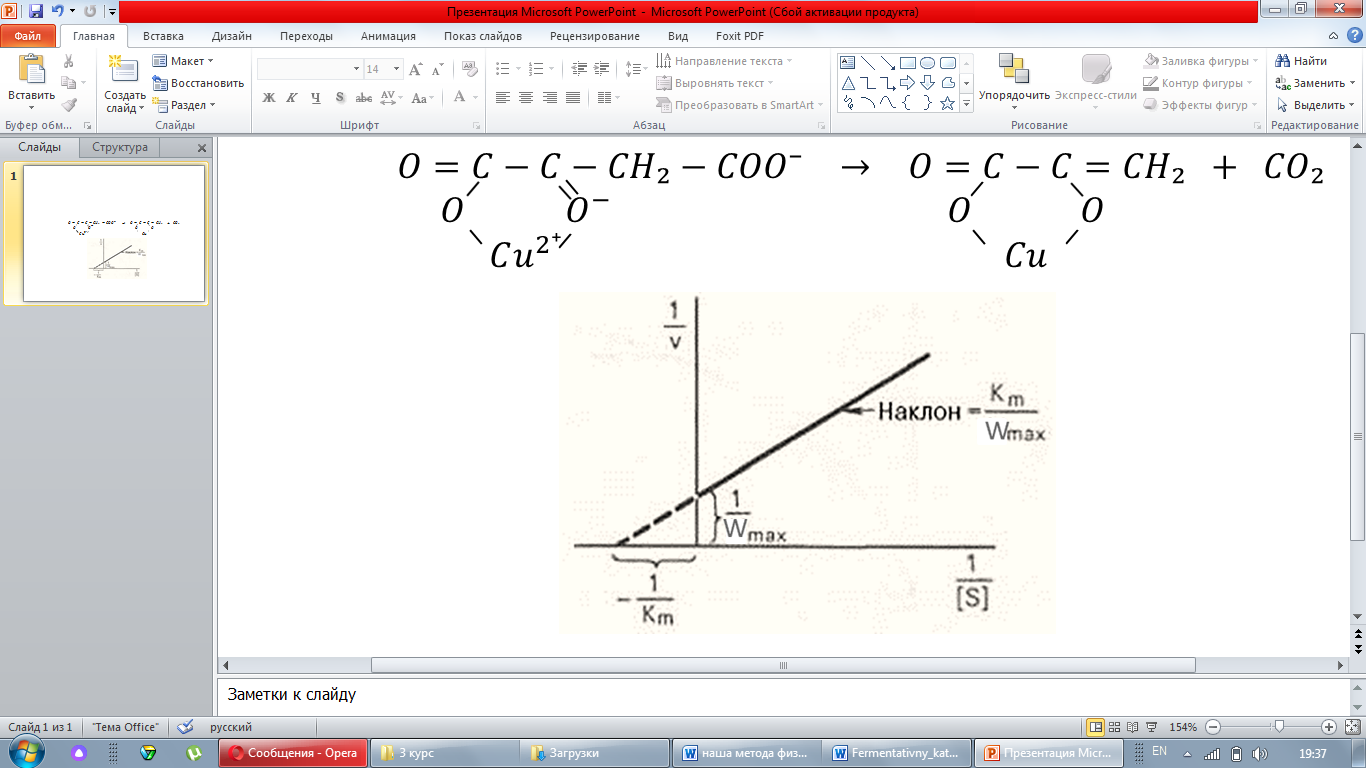
По этому графику Km и Wmax определяются более точно.