Материал: Задачи по биохимии
58. Ингибиторы синтеза тимидиловых нуклеотидов
Противоопухолевые препараты. Синтезировано очень много аналогов дНТФ, которые включаются ДНК полимеразами в ДНК и ингибируют репликацию. К числу мощных противоопухолевых препаратов принадлежит 5-фторурацил (5-FU) - аналог урацила.
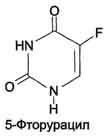
В организме по "запасным" путям 5-FU превращается в 5-Р-УМФ либо в реакции, катализируемой оротатфосфорибозилтрансферазой, либо через промежуточное образование нуклеозида и последующее фосфорилирование. Превращаясь в нуклеозиддифосфат, 5-FU может участвовать в реакции, катализируемой РНР, и восстанавливаться в соответствующее дезоксипроизводное. Под действием фосфатазы 5-Р-дУДФ снова теряет фосфат, и образующийся 5-Р-дУМФ связывается с тимидилатсинтазой и N5, N10-метилен-Н4-фолатом, образуя комплекс, напоминающий промежуточное соединение в реакции превращения дУМФ в дТМФ. Тимидилатсинтаза оказывается полностью блокированной, и синтез дТМФ прекращается.
Цитозинарабинозид (или цитарабин) представляет собой соединение, в котором остаток ри-бозы замещён на стериоизомер - арабинозу. Оно используется в химиотерапии рака, в частности, при острой миелоцитарной лейкемии. В организме препарат может превращаться в дНТФ, ингибировать ДНК полимеразы и снижать скорость репликации.
Аналоги фолиевой кислоты. В обмене нуклеотидов производные Н4-фолата как доноры од-ноуглеродных групп участвуют в формировании пуринового гетероциклического кольца и в ключевой реакции синтеза дТМФ из дУМФ, катализируемой тимидилатсинтазой.
В последнем случае N5,N10-метилен-Н4-фолат служит донором метальной группы и в ходе реакции превращается в Н2-фолат. Для активного синтеза тимидиловых нуклеотидов Н2-фолат должен повторно использоваться, проходя стадию восстановления в Н4-фолат (см. формулу ниже).
Метотрексат и аминоптерин - структурные аналоги фолиевой кислоты - ингибируют дигидрофолатредуктазу и таким образом нарушают синтез пуриновых нуклеотидов и превращение дУМФ в дТМФ, снижая внутриклеточную концентрацию субстратов синтеза ДНК и РНК. Препараты широко используют в химиотерапии опухолей.
Б. Антивирусные и антибактериальные препараты
Азидотимидин (AZT, или зидовидин) представляет собой мощный противовирусный препарат, применяющийся в лечении инфекций, которые сопровождают приобретённые формы иммунодефицита. Будучи структурным аналогом тимиди-на, препарат имеет в З'-положении дезоксирибозы азидогруппу (см. схему на с. 544).
AZT может фосфорилироваться и с помощью ДНК-полимераз включаться в растущую молекулу ДНК. Однако присутствие в 3'-положении дезоксирибозы азидогруппы делает синтезирующиеся молекулы ДНК не способными к последующему удлинению. В результате образование новых молекул ДНК прекращается.
Важно, что фосфорилированные производные AZT утилизируются более эффективно вирусной ДНК-полимеразой или так называемой обратной транскриптазой, чем ДНК-полимеразами эукариотов, поэтому препарат наиболее эффективно влияет на размножение вирусов и, в частности, ретровируса, вызывающего ВИЧ-инфекцию.
57. Патология пуринового и пиримидинового обмена: ТКИД (тяжелый комбинированный иммунодефицит), подагра, синдром Леша-Нихана, оротацидурия.
Тяжёлый комбинированный иммунодефицит- генетическое заболевание, при котором в результате дефекта одного из генов нарушается работа компонентов адаптивной иммунной системы B- и T-лимфоцитов. Тяжёлый комбинированный иммунодефицит— это тяжёлая форма наследственного иммунодефицита, который также известен как синдром мальчика в пузыре, так как больные крайне уязвимы перед инфекционными болезнями и вынуждены находиться в стерильной среде.
Симптомами тяжёлого комбинированного иммунодефицита могут являться хроническая диарея, ушные инфекции, возвратный пневмоцистоз, обильные кандидозы полости рта. Без лечения, в случае, если не было произведено успешной трансплантации гемопоэтических стволовых клеток, дети обычно умирают в течение первого года жизни от тяжёлых возвратных инфекций.
Существует несколько форм комбинированного иммунодефицита. Самая распространенная форма заболевания связана с мутацией гена X-хромосомы и встречается только у мужчин, так как они наследуют одну X-хромосому. Поскольку женщины наследуют две X-хромосомы (одну патологическую и одну нормальную), они являются только носителями заболевания, иммунные нарушения у них при этом отсутствуют.
Причиной другой формы заболевания является дефицит фермента аденозиндеаминазы, который необходим для расщепления пуринов. Недостаток аденозиндеаминазы провоцирует накопление dATP. Этот метаболит ингибирует активность фермента рибонуклеотидредуктазы, участвующего в превращении рибонуклеотидов в дезоксирибонуклеотиды. Если рибонуклеотидредуктаза не способна нормально функционировать, пролиферация лимфоцитов блокируется, а иммунная система компрометируется.
Остальные формы заболевания связаны с различными генетическими мутациями.
Подагра
Когда в плазме крови концентрация мочевой кислоты превышает норму, то возникает гиперурикемия. Вследствие гиперурикемии может развиться подагра - заболевание, при котором кристаллы мочевой кислоты и уратов откладываются в суставных хрящах, синовиальной оболочке, подкожной клетчатке с образованием подагрических узлов, или тофусов.
Биохимической основой развития некоторых форм подагры является нарушение катаболизма пуринов или гиперпродукция в тканях мочевой кислоты; в последнем случае генетический дефект состоит в аномально высокой активности фермента ФРПФ-синтетазы, который запускает каскад реакций биосинтеза пуринов.
С целью уменьшения гиперрурикемии при подагре предложен препарат Алопуринол,который по механизму действия является необратимым ингибитором ксантиноксидазы; применение препарата значительно уменьшает содержание мочевой кислоты в крови,что способствует облегчению клинических проявлений заболевания.
Синдром Леша-Нихана- тяжёлая форма гиперурикемии, которая наследуется как рецессивный признак, сцепленный с Х-хромосомой, и проявляется только у мальчиков.
Болезнь вызвана полным отсутствием активности гипоксантин-гуанинфосфорибозилтрансферазы - фермента, который обеспечивает повторное использование в метаболических реакциях свободных гипоксантина и гуанина – «путь реутилизации». Вследствие дефицита фермента в организме происходит аномальное накопление гипоксантина и гуанина, которые, превращаясь в мочевую кислоту, вызывают развитие гиперурикемии.
У детей с данной патологией в раннем возрасте появляются тофусы, уратные камни в моче-выводящих путях и серьёзные неврологические отклонения, сопровождающиеся нарушением речи, церебральными параличами, снижением интеллекта, склонностью к нанесению себе увечий (укусы губ, языка, пальцев).
В первые месяцы жизни неврологические расстройства не обнаруживаются, но на пелёнках отмечают розовые и оранжевые пятна, вызванные присутствием в моче кристаллов мочевой кислоты. При отсутствии лечения больные погибают в возрасте до 10 лет из-за нарушения функции почек.
Оротацидурия
Это единственное нарушение синтеза пиримидинов de novo. Оно вызвано снижением активности УМФ-синтазы, которая катализирует образование и декарбоксилирование ОМФ.
Установлено, что содержание оротовои кислоты в моче пациентов (1 г/сут и более) значительно превосходит количество оротата, которое ежедневно синтезируется в норме (около 600 мг/сут). Снижение синтеза пиримидиновых нуклеотидов, наблюдающееся при этой патологии, нарушает регуляцию КАД-фермента по механизму ретроингибирования, из-за чего возникает гиперпродукция оротата.
Клинически наиболее характерное следствие оротацидурии - мегалобластная анемия, вызванная неспособностью организма обеспечить нормальную скорость деления клеток эритроцитарного ряда. Её диагностируют у детей на том основании, что она не поддаётся лечению препаратами фолиевой кислоты.
Недостаточность синтеза пиримидиновых нуклеотидов сказывается на интеллектуальном развитии, двигательной способности и сопровождается нарушениями работы сердца и ЖКТ. Нарушается формирование иммунной системы, и наблюдается повышенная чувствительность к различным инфекциям.
Гиперэкскреция оротовои кислоты сопровождается нарушениями со стороны мочевыводящей системы и образованием камней. При отсутствии лечения больные обычно погибают в первые годы жизни. При этом оротовая кислота не оказывает токсического эффекта. Многочисленные нарушения в работе разных систем организма вызваны "пиримидиновым голодом".
Для лечения этой болезни применяют уридин, который по "запасному" пути превращается в УМФ.
Уридин + АТФ → УМФ + АДФ.
Нагрузка уридином устраняет "пиримидиновый голод", а поскольку из УМФ могут синтезироваться все остальные нуклеотиды пиримидинового ряда, то снижается выделение оротовой кислоты из-за восстановления механизма ретроингибирования КАД-фермента. Для больных оротацидурией лечение уридином продолжается в течение всей жизни, и этот нуклеозид становится для них незаменимым пищевым фактором.
59. Генетический код и его свойства.
Генетический код – система записи информации о последовательности расположения аминокислот в белках при помощи последовательности расположения нуклеотидов в ДНК.
Св-ва кода:
-
Триплетность-значащей единицей кода является сочетание трёх нуклеотидов (триплет, или кодон).
-
Специфичность – 1 кодон-1аминокислота.
-
Вырожденность (избыточность) — одной и той же аминокислоте может соответствовать несколько кодонов.
-
Универсальность — генетический код работает одинаково в организмах разного уровня сложности — от вирусов до человека (на этом основаны методы генной инженерии)
-
Однонаправленность – (5’ 3’)
-
Непрерывность - между триплетами нет знаков препинания, то есть информация считывается непрерывно.
-
Неперекрываемость — один и тот же нуклеотид не может входить одновременно в состав двух или более триплетов.
-
Линейность
Три кодона УАГ,УАА,УГА- не кодируют ни одной аминокислоты-СТОП кодон; СТАРТ кодон-АУГ(метионин)
73. Карликовость — аномально небольшой рост взрослого человека. Карликовость связана с недостатком гормона роста соматотропина или нарушением его конформации (строения). Причина: недостаток соматомединов, синтезирующихся в печени. Между соматомедином и соматотропинов имеется коррелляционная связь. Так, при пониженном синтезе соматомединов активность соматотропного гормона уменьшается, хотя функционирование гипофиза не нарушено.
Гигантизм (от др.-греч. — «великан, исполин, гигант») относится к заболеванию, обусловленному повышенной выработкой гормона роста (соматотропина) передней долей гипофиза, что приводит к чрезмерному пропорциональному росту туловища и конечностей и проявляющаяся уже в детском возрасте.
У больных, кроме того, наблюдается:
-
расстройство психического и физического состояния,
-
расстройство половой функции.
При гигантизме высок риск бесплодия и ограничена трудоспособность.
Акромегалия (от греч. ἄκρος — конечность и греч. μέγας — большой) — заболевание, связанное с нарушением функции передней доли гипофиза (аденогипофиз); сопровождается увеличением (расширением и утолщением) кистей,стоп, черепа, особенно его лицевой части, и др. Акромегалия возникает обычно после завершения роста организма; развивается постепенно, длится много лет. Вызывается выработкой чрезмерного количества соматотропного гормона. Аналогичное нарушение деятельности гипофиза в раннем возрасте вызывает гигантизм. При акромегалии отмечаются головные боли, утомляемость, ослабление умственных способностей, расстройство зрения, часто половое бессилие у мужчин и прекращение менструаций у женщин. Лечение — хирургическая операция на гипофизе, рентгенотерапия, применение гормональных препаратов, уменьшающих выработку СТГ (бромокриптин, ланреотид). Заболевание вызванной гипер продукцией СТГ. Почти у всех больных выявляются СТГ-секретирующие аденомы гипофиза, происходящие из соматотропных клеток. В подавляющем большинстве случаев опухолевую трансформацию этих клеток провоцируют активирующие мутации гена белка Gsальфа. Мутантный белок Gsальфа непрерывно стимулирует аденилатциклазу, что приводит к усилению пролиферации соматотропных клеток и к усилению продукции СТГ. В редких случаях гиперсекреция СТГ бывает вызвана гиперплазией гипофиза или избыточной секрецией соматолиберина.
|
|
|
|
Основные признаки заболевания:
-
Ожирение: жир откладывается на плечах, животе, лице, молочных железах и спине. Несмотря на тучное тело, руки и ноги у больных тонкие. Лицо становится лунообразным, круглым, щеки красными.
-
Розово-пурпурные или багровые полосы (стрии) на коже.
-
Избыточный рост волос на теле (у женщин растут усы и борода на лице).
-
У женщин — нарушение менструального цикла и бесплодие, у мужчин — снижение сексуального влечения и потенции.
-
Мышечная слабость.
-
Ломкость костей (развивается остеопороз), вплоть до патологических переломов позвоночника, ребер.
-
Повышается артериальное давление.
-
Нарушение чувствительности к инсулину и развитие сахарного диабета.
-
Возможно развитие мочекаменной болезни.
-
Иногда возникают нарушение сна, эйфория, депрессия.
-
Снижение иммунитета. Проявляется образованием трофических язв, гнойничковых поражений кожи, хронического пиелонефрита, сепсиса и т. д.
|
|
Несахарный диабет – заболевание, развивающееся при недостаточности выделения антидиуретического гормона (АДГ) или снижении чувствительности почечной ткани к его действию. |
74. Инсулин – это простой белок, который состоит из двух цепей: А (21 аминокислота) и В (30 аминокислот), связанных друг с другом двумя дисульфидными мостиками, третий S-S мостик находится в А-цепи. Вырабатывается в виде проинсулина, который состоит из одной полипептидной цепи (84 аминокислотных остатка), активируется путем частичного протеолиза. Синтез и секреция регулируются уровнем глюкозы в крови. Синтезируется бэта-клетками поджелудочной железы.
Секреция
Бета-клетки островков Лангерганса чувствительны к изменению уровня глюкозы в крови; выделение ими инсулина в ответ на повышение концентрации глюкозы реализуется по следующему механизму:
-
Глюкоза свободно транспортируется в бета-клетки специальным белком-переносчиком GluT 2.
-
В клетке глюкоза подвергается гликолизу и далее окисляется в дыхательном цикле с образованием АТФ; интенсивность синтеза АТФ зависит от уровня глюкозы в крови.
-
АТФ регулирует закрытие ионных калиевых каналов, приводя к деполяризации мембраны.
-
Деполяризация вызывает открытие потенциал-зависимых кальциевых каналов, это приводит к току кальция в клетку.
-
Повышение уровня кальция в клетке активирует фосфолипазу C, которая расщепляет один из мембранных фосфолипидов — фосфатидилинозитол-4,5-бифосфат — на инозитол-1,4,5-трифосфат и диацилглицерат.
-
Инозитолтрифосфат связывается с рецепторными белками ЭПР. Это приводит к высвобождению связанного внутриклеточного кальция и резкому повышению его концентрации.
-
Значительное увеличение концентрации в клетке ионов кальция приводит к высвобождению заранее синтезированного инсулина, хранящегося в секреторных гранулах.
В зрелых секреторных гранулах кроме инсулина и C-пептида находятся ионы цинка, амилин и небольшие количества проинсулина и промежуточных форм.
Выделение инсулина из клетки происходит путём экзоцитоза — зрелая секреторная гранула приближается к плазматической мембране и сливается с ней, и содержимое гранулы выдавливается из клетки. Изменение физических свойств среды приводит к отщеплению цинка и распаду кристаллического неактивного инсулина на отдельные молекулы, которые и обладают биологической активностью.
Механизм
Подобно другим гормонам своё действие инсулин осуществляет через белок-рецептор.
Инсулиновый рецептор представляет собой сложный интегральный белок клеточной мембраны, построенный из 2 субъединиц (a и b), причём каждая из них образована двумя полипептидными цепочками.
Инсулин с высокой специфичностью связывается и распознаётся а-субъединицей рецептора, которая при присоединении гормона изменяет свою конформацию. Это приводит к появлению тирозинкиназной активности у субъединицы b, что запускает разветвлённую цепь реакций по активации ферментов, которая начинается саутофосфорилирования рецептора.
Весь комплекс биохимических последствий взаимодействия инсулина и рецептора ещё до конца не вполне ясен, однако известно, что на промежуточном этапе происходит образование вторичных посредников: диацилглицеролов и инозитолтрифосфата, одним из эффектов которых является активация фермента —протеинкиназы С, с фосфорилирующим (и активирующим) действием которой на ферменты и связаны изменения во внутриклеточном обмене веществ.
Усиление поступления глюкозы в клетку связано с активирующим действием посредников инсулина на включение в клеточную мембрану цитоплазматических везикул, содержащих белок-переносчик глюкозы GLUT 4.
75. Глюкагон – одно цепочный полипептид, который состоит из 29 аминокислот, вырабатывается в виде проглюкагона (37 аминокислот), активируется частичным протеолизом, секреция стимулируется ионами кальция и аминокислотами, ингибируется глюкозой и соматостатином.