Материал: Ответы по ПА
Гидропическая дистрофия
Гидропическая, или водяночная, дистрофия характеризуется появлением в клетке вакуолей, наполненных цитоплазматической жидкостью. Она наблюдается чаще в эпителии кожи и почечных канальцев, в гепатоцитах, мышечных и нервных клетках, а также в клетках коры надпочечников.
Микроскопическая картина: паренхиматозные клетки увеличены в объеме, цитоплазма их заполнена вакуолями, содержащими прозрачную жидкость. Ядро смещается на периферию, иногда вакуолизируется или сморщивается. Прогрессирование этих изменений приводит к распаду ультраструктур клетки и переполнению клетки водой. Клетка превращается в заполненные жидкостью баллоны или в огромную вакуоль, в которой плавает пузырьковидное ядро. Такие изменения клетки, которые по существу являются выражением фокального колликвационного некроза называют баллонной дистрофией.
Внешний вид органов и тканей мало изменяется при гидропическои дистрофии, она обнаруживается обычно под микроскопом.
Механизм развития гидропическои дистрофии сложен и отражает нарушения водно-электролитного и белкового обмена, ведущие к изменению коллоидно-осмотического давления в клетке. Большую роль играет нарушение проницаемости мембран клетки, сопровождающееся их распадом. Это ведет к закислению цитоплазмы, активации гидролитических ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды.
Причины развития гидропической дистрофии в разных органах неоднозначны. В почках - это повреждение гломерулярного фильтра (гломерулонефрит, амилоидоз, сахарный диабет), что ведет к гиперфильтрации и недостаточности ферментной системы базального лабиринта нефроцитов, в норме обеспечивающей реабсорбцию воды; поэтому гидропическая дистрофия нефроцитов так характерна для нефротического синдрома. В печени гидропическая дистрофия возникает при вирусном и токсическом гепатитах и нередко является причиной печеночной недостаточности. Причиной гидропическои дистрофии эпидермиса может быть инфекция (оспа), отек кожи различного механизма. Вакуолизация цитоплазмы может быть проявлением физиологической деятельности клетки, что отмечается, например, в ганглиозных клетках центральной и периферической нервной системы.
Исход гидропической дистрофии, как правило, неблагоприятный; она завершается фокальным или тотальным некрозом клетки. Поэтому функция органов и тканей при гидропической дистрофии резко страдает
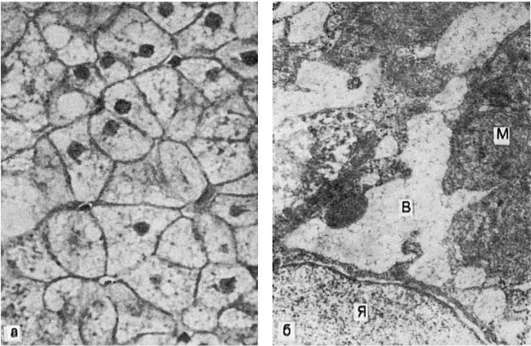
Гидропическая дистрофия печени (биопсия):
а - микроскопическая картина; вакуолизация гепатоцитов; б - электронограмма: рас- ширение канальцев эндоплазматической сети и образование вакуолей (В), заполненных хлопьевидным содержимым. Мембраны, ограничивающие вакуоли, почти полностью ли- шены рибосом. Вакуоли сдавливают расположенные между ними митохондрии (М), часть которых подвергается деструкции; Я - ядро гепатоцита. х18 000
Роговая дистрофия
Роговая дистрофия, или патологическое ороговение, характеризуется избыточным образованием рогового вещества в ороговевающем эпителии (гиперкератоз, ихтиоз) или образованием рогового вещества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках, или лейкоплакия; образование «раковых жемчужин» в плоскоклеточном раке). Процесс может быть местным или распространенным.
Причины роговой дистрофии разнообразны: нарушение развития кожи, хроническое воспаление, вирусные инфекции, авитаминозы и др.
Исход может быть двояким: устранение вызывающей причины в начале процесса может привести к восстановлению ткани, однако в далеко зашедших случаях наступает гибель клеток.
Значение роговой дистрофии определяется ее степенью, распространенностью и длительностью. Длительно существующее патологическое ороговение слизистой оболочки (лейкоплакия) может явиться источником развития раковой опухоли. Врожденный ихтиоз резкой степени, как правило, несовместим с жизнью.
9.Внутриклеточные накоплениягликогена: причины, патогенез, морфогенез, морфологическая характеристика и методы диагностики.
Углеводные дистрофии, связанные с нарушением обмена гликогена
Основные запасы гликогена находятся в печени и скелетных мышцах. Гликоген печени и мышц расходуется в зависимости от потребностей организма (лабильный гликоген). Гликоген нервных клеток, проводящей системы сердца, аорты, эндотелия, эпителиальных покровов, слизистой оболочки матки, соединительной ткани, эмбриональных тканей, хряща и лейкоцитов является необходимым компонентом клеток, и его содержа- ние не подвергается заметным колебаниям (стабильный гликоген). Однако деление гликогена на лабильный и стабильный условно.
Регуляция обмена углеводов осуществляется нейроэндокринным путем. Основная роль принадлежит гипоталамической области, гипофизу (АКТГ, тиреотропный, соматотропный гормоны), (β-клеткам поджелудочной железы (инсулин), надпочечникам (глюкокортикоиды, адреналин) и щитовидной железе.
Нарушения содержания гликогена проявляются в уменьшении или увеличении количества его в тканях и появлении там, где он обычно не выявляется. Эти нарушения наиболее ярко выражены при сахарном диабете и при наследственных углеводных дистрофиях - гликогенозах.
При сахарном диабете, развитие которого связывают с патологией β-клеток островков поджелудочной железы, происходят недостаточное использование глюкозы тканями, увеличение ее содержания в крови (гипергликемия) и выведение с мочой (глюкозурия). Тканевые запасы гликогена резко уменьшаются. Это в первую очередь касается печени,в которой нарушается синтез гликогена, что ведет к инфильтрации ее жирами - развивается жировая дистрофия печени; при этом в ядрах гепатоцитов появляются включения гликогена, они становятся светлыми («дырчатые», «пустые», ядра).
С глюкозурией связаны характерные изменения почек при диабете. Они выражаются в гликогенной инфильтрации эпителия канальцев, главным образом узкого и дистального сегментов. Эпителий становится высоким, со светлой пенистой цитоплазмой; зерна гликогена видны и в просвете канальцев. Эти изменения отражают состояние синтеза гликогена (полимеризация глюкозы) в канальцевом эпителии при резорбции богатого глюкозой ультрафильтрата плазмы.
При диабете страдают не только почечные канальцы, но и клубочки, их капиллярные петли, базальная мембрана которых становится значительно более проницаемой для сахаров и белков плазмы. Возникает одно из проявлений диабетической микроангиопатии - интеркапиллярный (диабетический) гломерулосклероз.
Наследственные углеводные дистрофии, в основе которых лежат нарушения обмена гликогена, называются гликогенозами. Гликогенозы обусловлены отсутствием или недостаточностью фермента, участвующего в расщеплении депонированного гликогена, и относятся поэтому к наследственным ферментопатиям, или болезням накопления. В настоящее время хорошо изучены 6 типов гликогенозов, обусловленных наследственной недостаточностью 6 различных ферментов. Это болезни Гирке (I тип), Помпе (II тип), Мак-Ардля (V тип) и Герса (VI тип), при которых структура накапливаемого в тканях гликогена не нарушена, и болезни Форбса-Кори (III тип) и Андерсена (IV тип), при которых она резко изменена
Морфологическая диагностика гликогеноза того или иного типа возможна при биопсии с помощью гистоферментативных методов.
10.Нарушения обмена липофусцина и меланина: клинико-морфологическая характеристика.
Нарушения обмена хромопротеидов (эндогенные пигментации)2
Хромопротеиды - окрашенные белки, или эндогенные пигменты, играют важную роль в жизни организма. С помощью хромопротеидов осуществляются дыхание (гемоглобин, цитохромы), выработка секретов (желчь) и инкретов (серотонин), защита организма от воздействия лучевой энергии (меланин), пополнение запасов железа (ферритин), баланс витаминов (липохромы) и т.д. Обмен пигментов регулируется вегетативной нервной системой, эндокринными железами, он тесно связан с функцией органов кроветворения и системы моноцитарных фагоцитов.
Классификация. Эндогенные пигменты принято делить на 3 группы:
-гемоглобиногенные, представляющие собой различные производные гемоглобина
-протеиногенные, или тирозиногенные, связанные с обменом тирозина
- липидогенные, или липопигменты, образующиеся при обмене жиров.
Нарушения обмена протеиногенных (тирозиногенных) пигментов
К протеиногенным (тирозиногенным) пигментам относят меланин, пигмент гранул энтерохромаффинных клеток и адренохром. Накопление этих пигментов в тканях служит проявлением ряда заболеваний.
Меланин (от греч. melas - черный) - широко распространенный буро-черный пигмент, с которым у человека связана окраска кожи, волос, глаз. Он дает положительную аргентаффинную реакцию, т.е. обладает способностью восстанавливать аммиачный раствор нитрата серебра до металлического серебра. Эти реакции позволяют гистохимически отличить его в тканях от других пигментов.
Синтез меланина происходит из тирозина в клетках меланинобразующей ткани - меланоцитах. Их предшественниками являются меланобласты. Под действием тирозиназы в меланосомах меланоцитов из тирозина образуется диоксифенилаланин (ДОФА), или промеланин, который полимеризуется в меланин. Клетки, фагоцитирующие меланин, называют меланофагами.
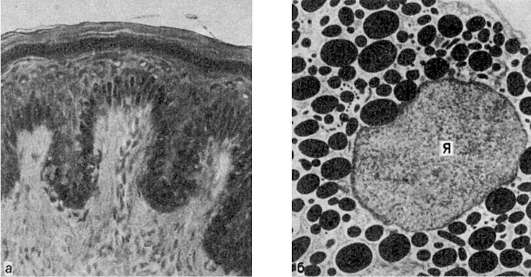
Кожа при аддисоновой болезни:
а - в базальном слое эпидермиса скопления меланоцитов; в дерме много меланофагов; б - меланоцит кожи. В цитоплазме много меланосом. Я - ядро. Электронограмма. х10 000
Меланоциты и меланофаги содержатся в эпидермисе, дерме, радужной и сетчатой оболочках глаз, в мягкой мозговой оболочке. Содержание меланина в коже, сетчатке и радужке зависит от индивидуальных и расовых особенностей и подвергается колебаниям в различные периоды жизни. Регуляция меланогенеза осуществляется нервной системой и эндокринными железами. Стимулируют синтез меланина меланостимулирующий гормон гипофиза, АКТГ, половые гормоны, медиаторы симпатической нервной системы, тормозят - мелатонин и медиаторы парасимпатической нервной системы. Образование меланина стимулируется ультрафиолетовыми лучами, что объясняет возникновение загара как адаптивной защитной биологической реакции.
Нарушения обмена меланина выражаются в усиленном его образовании или исчезновении. Эти нарушения имеют распространенный или местный характер и могут быть приобретенными или врожденными.
Распространенный приобретенный гипермеланоз (меланодермия) особенно часто и резко выражен при аддисоновой болезни , обусловленной поражением надпочечников, чаще туберкулезной или опухолевой природы. Гиперпигментация кожи при этой болезни объясняется не столько тем, что при разрушении надпочечников вместо адреналина из тирозина и ДОФА синтезируется меланин, сколько усилением продукции АКТГ в ответ на уменьшение адреналина в крови. АКТГ стимулирует синтез меланина, в меланоцитах увеличивается количество меланосом. Меланодермия встречается также при эндокринных расстройствах (гипогонадизм, гипопитуитаризм), авитаминозах (пеллагра, цинга), кахексии, интоксикации углеводородами.
Распространенный врожденный гипермеланоз (пигментная ксеродерма) связан с повышенной чувствительностью кожи к ультрафиолетовым лучам и выражается в пятнистой пигментации кожи с явлениями гиперкератоза и отека.
К местному приобретенному меланозу относят меланоз толстой кишки, который встречается у людей, страдающих хроническим запором, гиперпигментированные участки кожи (черный акантоз) при аденомах гипофиза, гипертиреоидизме, сахарном диабете. Очаговое усиленное образование меланина наблюдается в пигментных пятнах (веснушки, лентиго) и в пигментных невусах. Из пигментных невусов могут возникать злокачественные опухоли - меланомы.
Распространенный гипомеланоз, или альбинизм (от лат. albus - белый), связан с наследственной недостаточностью тирозиназы. Альбинизм проявляется отсутствием меланина в волосяных луковицах, эпидермисе и дерме, в сетчатке и радужке.
Очаговый гипомеланоз (лейкодерма, или витилиго) возникает при нарушении нейроэндокринной регуляции меланогенеза (лепра, гиперпаратиреоидизм, сахарный диабет), образовании антител к меланину (зоб Хасимото), воспалительных и некротических поражениях кожи (сифилис).
Нарушения обмена липидогенных пигментов (липопигментов)
В эту группу входят жиробелковые пигменты - липофусцин, пигмент недостаточности витамина Е, цероид и липохромы. Липофусцин, пигмент недостаточности витамина Е и цероид имеют одинаковые физические и химические (гистохимические) свойства, что дает право считать их разновидностями одного пигмента - липофусцина. Однако в настоящее время липофусцином считают липопигмент лишь паренхиматозных и нервных клеток; пигмент недостаточности витамина Е - разновидность липофусцина. Цероидом называют липопигмент мезенхимальных клеток, главным образом макрофагов.
Патология обмена липопигментов разнообразна.
Липофусцин представляет собой гликолипопротеид. Он представлен зернами золотистого или коричневого цвета, электронно-микроскопически выявляется в виде электронно-плотных гранул , окруженных трехконтурной мембраной, которая содержит миелиноподобные структуры.
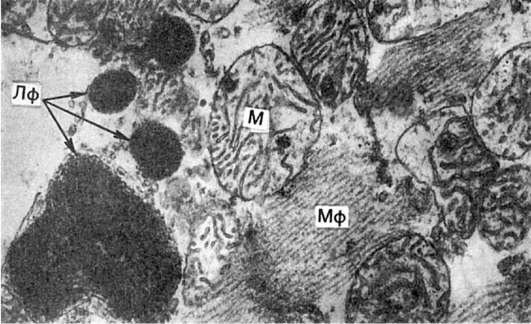
Липофусцин (Лф) в мышечной клетке сердца, тесно связанный с митохондриями (М). Мф - миофибриллы. Электронограмма. х21 000
Образование липофусцина происходит путем аутофагии и проходит несколько стадий. Первичные гранулы, или пропигмент-гранулы, появляются перинуклеарно в зоне наиболее активно протекающих обменных процессов. Они содержат ферменты митохондрий и рибосом (металлофлавопротеиды, цитохромы), связанные с липопротеидами их мембран. Пропигмент-гранулы поступают в пластинчатый комплекс, где происходит синтез гранул незрелого липофусцина, который суданофилен, ШИК-положителен, содержит железо, иногда медь, обладает светло-желтой аутофлюоресценцией в ультрафиолетовом свете. Гранулы незрелого пигмента перемещаются в периферическую зону клетки и абсорбируются там лизосомами; появляется зрелый липофусцин, обладающий высокой активностью лизосомных, а не дыхательных ферментов. Гранулы его становятся коричневыми, они стойко суданофильны, ШИК-положительны, железо в них не выявляется, аутофлюоресценция становится красно-коричневой. Накапливающийся в лизосомах липофусцин превращается в остаточные тельца - телолизосомы.
В условиях патологии содержание липофусцина в клетках может резко увеличиваться. Это нарушение обмена называется липофусцинозом. Он может быть вторичным и первичным (наследственным).
Вторичный липофусциноз развивается в старости, при истощающих заболеваниях, ведущих к кахексии (бурая атрофия миокарда, печени), при повышении функциональной нагрузки (липофусциноз миокарда при пороке сердца, печени - при язвенной болезни желудка и двенадцатиперстной кишки), при злоупотреблении некоторыми лекарствами (анальгетики), при недостаточности витамина Е (пигмент недостаточности витамина Е).
Первичный (наследственный) липофусциноз характеризуется избирательным накоплением пигмента в клетках определенного органа или системы. Он проявляется в виде наследственного гепатоза, или доброкачественной гипербилирубинемии (синдромы Дабина-Джонсона, Жиль- бера, Кригера-Найяра) с избирательным липофусцинозом гепатоцитов, а также нейронального липофусциноза (синдром Бильшовского-Янского, Шпильмейера-Шегрена, Кафа), когда пигмент накапливается в нервных клетках, что сопровождается снижением интеллекта, судорогами, нарушением зрения.
11.Гемосидероз местный и системный. Гемохроматоз. Морфологическая характеристика и методы диагностики.
Нарушения обмена гемоглобиногенных пигментов
В норме гемоглобин проходит ряд циклических превращений, обеспечивающих его ресинтез и образование необходимых для организма продуктов. Эти превращения связаны со старением и разрушением эритроцитов (гемолиз, эритрофагия), постоянным обновлением эритроцитной массы. В результате физиологического распада эритроцитов и гемоглобина образуются пигменты ферритин, гемосидерин и билирубин. В патологических условиях вследствие многих причин гемолиз может быть резко усилен и осуществляться как в циркулирующей крови (интраваскулярно), так и в очагах кровоизлияний (экстраваскулярно). В этих условиях, помимо увеличения образующихся в норме гемоглобиногенных пигментов, может появляться ряд новых пигментов - гематоидин, гематины и порфирин.
В связи с накоплением гемоглобиногенных пигментов в тканях могут возникать различные виды эндогенных пигментации, которые становятся проявлением ряда заболеваний и патологических состояний.
Ферритин - железопротеид, содержащий до 23% железа. Железо ферритина связано с белком, который носит название апоферритина. В норме ферритин обладает дисульфидной группой. Это неактивная (окисленная) форма ферритина - SS-ферритин. При недостаточности кислорода происходит восстановление ферритина в активную форму - SH-ферритин, который обладает вазопаралитическими и гипотензивными свойствами. В зависимости от происхождения различают анаболический и катаболический ферритин. Анаболический ферритин образуется из железа, всасывающегося в кишечнике, катаболический - из железа гемолизированных эритроцитов. Ферритин (апоферритин) обладает антигенными свойствами. Ферритин образует берлинскую лазурь (железосинеродистое железо) под действием железосинеродистого калия и соляной, или хлористоводородной, кислоты (реакция Перлса) и может быть идентифицирован с помощью специфической антисыворотки при иммунофлюоресцентном исследовании. Большое количество ферритина содержится в печени (депо ферритина), селезенке, костном мозге и лимфатических узлах, где обмен его связан с синтезом гемосидерина, гемоглобина и цитохромов.
В условиях патологии количество ферритина может увеличиваться как в тканях, так и в крови. Повышение содержания ферритина в тканях наблюдается при гемосидерозе, так как полимеризация ферритина ведет к образованию гемосидерина. Ферритинемией объясняют необратимость шока, сопровождающегося сосудистым коллапсом, так как SH-ферритин выступает в роли антагониста адреналина.
Гемосидерин образуется при расщеплении гема и является полимером ферритина. Он представляет собой коллоидную гидроокись железа, связанную с белками, гликозаминогликанами и липидами клетки. Клетки, в которых образуется гемосидерин, называются сидеробластами. В их сидеросомах происходит синтез гранул гемосидерина . Сидеробласты могут быть как мезенхимальной, так и эпителиальной природы. Гемосидерин постоянно обнаруживается в ретикулярных и эндотелиальных клетках селезенки, печени, костного мозга, лимфатических узлах. В межклеточном веществе он подвергается фагоцитозу сидерофагами.
Присутствие в гемосидерине железа позволяет выявлять его с помощью характерных реакций: образование берлинской лазури (реакция Перлса), турнбулевой сини (обработка срезов сульфидом аммония, а затем железо- синеродистым калием и хлористово- дородной кислотой). Положительные реакции на железо отличают гемосидерин от сходных с ним пигментов (гемомеланин, липофусцин, меланин).
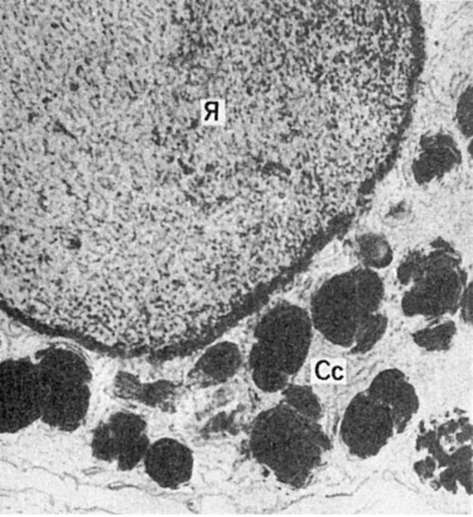
Сидеробласт. Крупное ядро (Я), узкий ободок цитоплазмы с большим числом сидеросом (Сс). Электронограмма. х20 000
В условиях патологии наблюдается избыточное образование гемосидерина - гемосидероз. Он может иметь как общий, так и местный характер.
Общий, или распространенный, гемосидероз наблюдается при внутрисосудистом разрушении эритроцитов (интраваскулярный гемолиз) и встречается при болезнях системы кроветворения (анемии, гемобластозы), интоксикациях гемолитическими ядами, некоторых инфекционных заболеваниях (возвратный тиф, бруцеллез, малярия и др.), переливаниях иногруппной крови, резус-конфликте и т.д. Разрушенные эритроциты, их обломки, гемоглобин идут на построение гемосидерина. Сидеробластами становятся ретикулярные, эндотелиальные и гистиоцитарные элементы селезенки, печени, костного мозга, лимфатических узлов, а также эпителиальные клетки печени, почек, легких, потовых и слюнных желез. Появляется большое число сидерофагов, которые не успевают поглощать гемосидерин, загружающий межклеточное вещество. В результате этого коллагеновые и эластические волокна пропитываются железом. При этом селезенка, печень, костный мозг и лимфатические узлы становятся ржаво-коричневыми.