Материал: Internal_diseases_propedeutics._Part_II._Diagnostics_of_cardiovascular_diseases
include increased cardiac work, reduced coronary perfusion, increased cardiac preload and afterload, fluid retention resulting in congestion, myocyte loss, increased K excretion, and cardiac arrhythmia.
Renal responses: The mechanism by which an asymptomatic patient with cardiac dysfunction develops overt CHF is unknown, but it begins with renal retention of Na and water, secondary to decreased renal perfusion. Thus, as cardiac function deteriorates, renal blood flow decreases in proportion to the reduced CO, the GFR falls, and blood flow within the kidney is redistributed. The filtration fraction and filtered Na decrease, but tubular resorption increases.
Neurohumoral responses: Increased activity of the renin-angiotensin-aldosterone system influences renal and peripheral vascular response in HF. The intense sympathetic activation accompanying HF stimulates the release of renin from the juxtaglomerular apparatus near the descending loop of Henle in the kidney. Probably, decreased arterial systolic stretch secondary to declining ventricular function also stimulates renin secretion. Reflex and adrenergic stimulation of the renin-angiotensin-aldosterone system produces a cascade of potentially deleterious effects: Increased aldosterone levels enhance Na reabsorption in the distal nephron, contributing to fluid retention. Renin produced by the kidney interacts with angiotensinogen, producing angiotensin I from which is cleaved the octapeptide angiotensin II by ACE. Angiotensin II has various effects believed to enhance the syndrome of CHF, including stimulation of the release of arginine vasopressin (AVP), which is antidiuretic hormone (ADH); vasoconstriction; enhanced aldosterone output; efferent renal vasoconstriction; renal Na retention; and increased norepinephrine release. Angiotensin II is also believed to be involved in vascular and myocardial hypertrophy, thus contributing to the remodeling of the heart and peripheral vasculature, which contributes to HF in various myocardial and other heart diseases.
Plasma norepinephrine levels are markedly increased, largely reflecting intense sympathetic nerve stimulation, because plasma epinephrine levels are not increased. High plasma norepinephrine levels in patients with CHF are associated with a poor prognosis.
The heart contains many neurohormonal receptors (α1, β1, β2, β3, adrenergic, muscarinic, endothelin, serotonin, adenosine, angiotensin II). In patients with HF, β1
56
receptors (which constitute 70% of cardiac β receptors), but not the other adrenergic receptors, are down-regulated, potentially adversely affecting myocardial function. This down-regulation, which is probably a response to intense sympathetic overdrive, has been detected even in asymptomatic patients with the early stages of HF. Altered myocardial stimulator or receptor functions for various other neurohormonal factors may adversely influence myocyte performance in HF.
Serum levels of atrial natriuretic peptide (released in response to increased atrial volume and pressure load) and brain natriuretic peptide (released from the ventricle in response to ventricular stretch) are markedly increased in patients with CHF. These peptides enhance renal excretion of Na, but, in patients with CHF, the effect is blunted by decreased renal perfusion pressure, receptor down-regulation, and perhaps enhanced enzymatic degradation. Serum, brain (B-type) natriuretic peptide appears to be important for diagnosis in CHF and correlates well with functional impairment.
AVP is released in response to a fall in BP or ECF volume and by the effects of various neurohormonal stimuli. An increase in plasma AVP diminishes excretion of free water by the kidney and may contribute to the hyponatremia of HF. AVP levels in CHF vary, but experimental AVP blockers have increased water excretion and serum Na levels.
Other consequences: Protein-losing enteropathy characterized by marked hypoalbuminemia, ischemic bowel infarction, acute and chronic GI hemorrhage, and malabsorption may result from severe chronic venous hypertension. Peripheral gangrene in the absence of large vessel occlusion or chronic irritability and decreased mental performance may result from chronic markedly reduced PO2, reflecting severely reduced cerebral blood flow and hypoxemia.
Cardiac cachexia (loss of lean tissue >= 10%) may accompany severely symptomatic HF. The failing heart produces tumor necrosis factor-, which is a key cytokine in the development of catabolism and possibly of cardiac cachexia. Marked anorexia is characteristic of this syndrome. Restoring cardiac function to normal can reverse cardiac cachexia.
Symptoms and Signs
57
HF may be predominantly right-sided or left-sided and may develop gradually or suddenly (as with acute pulmonary edema).
Cyanosis may occur with any form of HF. The cause may be central and may reflect hypoxemia. A peripheral component due to capillary stasis with increased A-VO2 and resultant marked venous oxyhemoglobin unsaturation may also be present. Improved color of the nail bed with vigorous massage suggests peripheral cyanosis. Central cyanosis cannot be altered by increasing local blood flow.
LV failure: Pulmonary venous hypertension may become apparent with tachycardia, fatigue on exertion, dyspnea on mild exercise, and intolerance to cold. Paroxysmal nocturnal dyspnea and nocturnal cough reflect the redistribution of excess fluid into the lung with the recumbent position. Occasionally, pulmonary venous hypertension and increased pulmonary fluid manifest as bronchospasm and wheezing. Cough may be prominent, and pink-tinged or brownish sputum due to blood and the presence of HF cells is common. Frank hemoptysis due to ruptured pulmonary varices with massive blood loss is uncommon but may occur. Signs of chronic LV failure include diffuse and laterally displaced apical impulse, palpable and audible ventricular (S3) and atrial gallops (S4), accentuated pulmonic second sound, and inspiratory basilar rales (crepitation). Rightsided pleural effusion is common.
Acute pulmonary edema is a life-threatening manifestation of acute LV failure secondary to sudden onset of pulmonary venous hypertension. A sudden rise in LV filling pressure results in rapid movement of plasma fluid through pulmonary capillaries into the interstitial spaces and alveoli. The patient presents with extreme dyspnea, deep cyanosis, tachypnea, hyperpnea, restlessness, and anxiety with a sense of suffocation. Pallor and diaphoresis are common. The pulse may be thready (pulsus filiformis), and BP may be difficult to obtain. Respirations are labored, and moist bubbling rales are widely dispersed over both lung fields anteriorly and posteriorly. Some patients manifest marked bronchospasm or wheezing (cardiac asthma). Noisy respiratory efforts often render cardiac auscultation difficult, but a summation gallop, merger of S3 and S4, may be heard. Hypoxemia is severe. CO2 retention is a late, ominous manifestation of secondary hypoventilation and requires immediate attention.
58
RV failure: The principal symptoms include fatigue; awareness of fullness in the neck; fullness in the abdomen, with occasional tenderness in the right upper quadrant (over the liver); ankle swelling; and, in advanced stages, abdominal swelling due to ascites. Edema over the sacrum is likely in supine patients. Signs include evidence of systemic venous hypertension, abnormally large a or v waves in the external jugular pulse, an enlarged and tender liver, a murmur of tricuspid regurgitation along the left sternal border, RV S3 and S4, and pitting edema of the lowest parts of the body Diagnosis
Although symptoms and signs (eg, exertional dyspnea, orthopnea, edema, tachycardia, pulmonary rales, a third heart sound, jugular venous distention) have a diagnostic specificity of 70 to 90%, the sensitivity and predictive accuracy are low.
Elevated levels of B-type natriuretic peptide are diagnostic. Adjunctive tests include CBC, blood creatinine, electrolytes (eg, Mg, Ca), glucose, albumin, and liver function tests. Thyroid function test results should be assessed in patients with atrial fibrillation and in selected, especially older, persons. In patients with suspected coronary artery disease, stress testing with radionuclide or ultrasound imaging or coronary angiography may be indicated. Endocardial biopsy is of limited usefulness.
ECG should be performed in all patients with HF, although findings are not specific; ambulatory ECG is not generally useful. Various abnormalities (eg, of ventricular hypertrophy, MI, or bundle branch block) may provide etiologic clues. Recent onset of rapid atrial fibrillation may precipitate acute LV or RV failure. Frequent premature ventricular contractions may be secondary and may subside when the HF is treated.
Chest x-ray should be performed in all patients (Fig.25 &26). Pulmonary venous congestion and interstitial or alveolar edema are characteristics of pulmonary edema. Kerley B lines reflect chronic elevation of left atrial pressure and chronic thickening of the intralobular septa from edema.
59
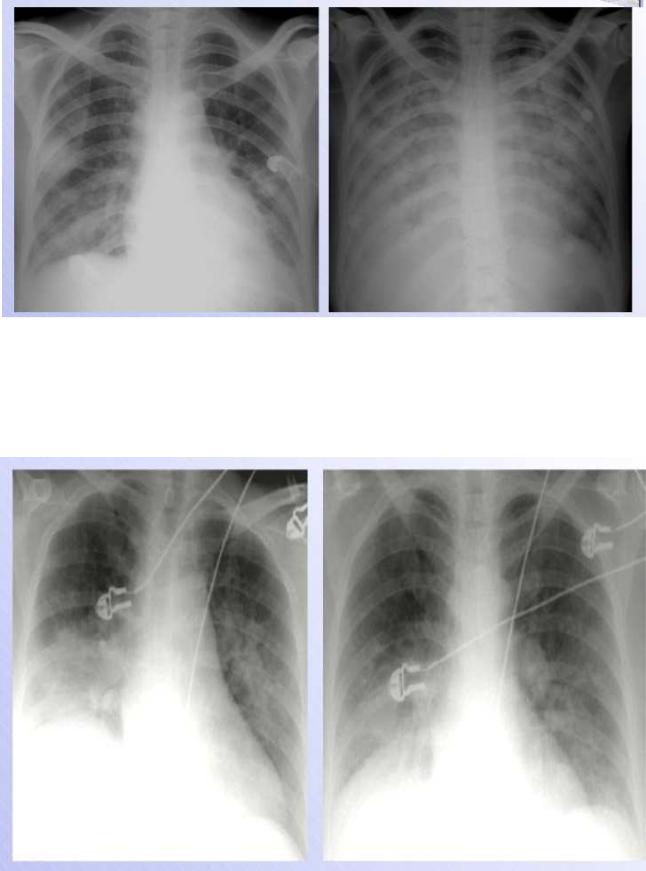
Fig.25. Heart failure
The upper zone bloo d vessels are distended and there are li near densities in the periphery of the lower z ones (interstitial or Kerley‘s B lines) a nd there are some areas of apparent co nsolidation, indicating alveolar pulmonary edema.
Fig.26 H eart failure following myocardial infarction.
60