Материал: 6. Лекция. Обмен холестерола, желчных кислот и кетоновых тел
Энергетический эффект окисления β-гидроксибутирата:
3 АТФ + 24 АТФ – 1 АТФ (затрачивается на активацию) = 26 АТФ
НАДН+Н+ 2 молекулы ацетил-SКоА
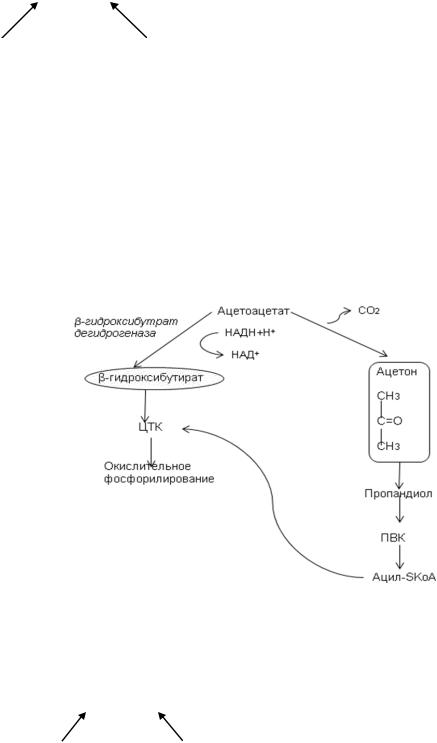
Помимо этого, ацетоацетат может восстанавливаться в β-гидроксибутират
(фермент - β-гидроксибутиратдегидрогеназа, кофактор НАДН++Н+). Это обратимая реакция. Ацетоацетат также может подвергаться нефементативному декарбоксилированию с образованием ацетона. Ацетон затем превращается в пропандиол, из которого синтезируется ПВК. Из ПВК в ходе окислительного декарбоксилирования образуется ацетилSKoA,
который включается в ЦТК.
Метаболизм ацетоацетата в мозговой ткани при голодании.
Энергетический эффект окисления ацетоацетата: 3 АТФ + 12 АТФ = 15 АТФ
НАДН+Н+ 2 ацетилSКоА
(окислительное декарбоксилирование ПВК).
Регуляция обмена кетоновых тел
Первый пункт координации кетогенеза – это уровень свободных жирных кислот в крови. Источником ВЖК является липолиз ТАГ в жировой ткани.
Печень захватывает 30% ВЖК, циркулирующих в крови, в результате чего повышается их концентрация в печёночной ткани. Это приводит к блокированию биосинтеза жирных кислот за счёт ингибирования ацетил- KoA-карбоксилазы (регуляторный фермент биосинтеза ЖК), а значит, и
снижению уровня малонил-KoA.
Второй пункт координации кетогенеза – митохондриальная ацил-карнитин транспортная система (перенос ацил-КоА в матрикс митохондрий).
Регулируемым ферментом данной системы является ацил-
карнитинтрансфераза I, а его отрицательным модулятором является малонилKoA. Снижение концентрации малонил-KoA снимает блок с ацил-
карнитинтрансферазы I и ацил-КоА поступает в матрикс митохондрий, где подвергается β-окислению с образованием большого количества ацетил-KoA.
Третий пункт координации кетогенеза – скорость ЦТК, которую регулирует фермент цитратсинтаза. Для осуществления ЦТК необходима высокая концентрация оксалоацетата (ОАА). При дефиците углеводов (глюкозы)
уровень ОАА уменьшается, так как он является субстратом глюконеогенеза.
В условиях дефицита ОАА уменьшается скорость синтеза цитрата, что приводит к накоплению ацетил-KoA. В матриксе митохондрий ацетил-KoA
используется в синтезе кетоновых тел. Поэтому можно сказать, что «жиры сгорают в пламени углеводов».
Печень не использует кетоновые тела, т.к. в ней отсутствуют ферменты их утилизации. Поэтому кетоновые тела поступают в кровь и транспортируются к органам и тканям, которые используют их в качестве энергетических субстратов. Сердечная мышца и корковый слой почек предпочтительно используют в качестве энергетического субстрата ацетоацетат, а не глюкозу.
В противоположность этому глюкоза является главным субстратом окисления для мозга у лиц, получающих сбалансированную пищу. При голодании и диабете мозг адаптируется к кетоновым телам.
Кетонемия – это повышение уровня кетоновых тел в крови. При патологических состояниях (сахарный диабет, голодание) концентрация
кетоновых тел в сыворотке крови увеличивается до 16-20 ммоль/л (в норме
0,03-0,2 ммоль/л). Кетонемия может привести к нарушению КОС крови. В
результате развивается кетоацидоз (метаболический ацидоз). 90% β-
гидроксибутирата находится в ионизированном состоянии. Следовательно,
увеличивается количество Н+ (протонов). В результате снижается рН крови:
СН3 – СН – СН2 – СООН ↔ СН3 – СН – СН2 – СОО- + Н+
│ |
│ |
ОН |
ОН |
Кетонурия – это появление кетоновых тел в моче. Кетоновые тела относятся к патологическим компонентам мочи.
Для коррекции кетоацидоза организм запускает защитные механизмы.
Первая линия защиты КОС – буферные системы крови
1. Бикарбонатная буферная система: [НСО3]/[CO2] = 20/1; Н+ + НСО3 →Н2СО3
Следовательно, увеличивается парциальное давление СО2.
Следующая система, которая начинает работать, это:
1.Гемоглобиновая буферная система:
СО2
Эритроцит СО2 + Н2О → Н2СО3
эффект
Н2СО3 + НbО2- Бора О2 + ННb + НСО3-
в кровь
Буферные системы крови
Второй линией защиты являются лёгкие и почки.
Через лёгкие удаляется избыток СО2. В почках повышается реабсорбция бикарбоната и кислые продукты – кетоновые тела - выводятся с мочой. 50%
кетоновых тел удаляется в молекулярном виде (сами протонируются),
остальные 50% удаляется в комплексе с щелочными металлами (Na+, K+).
В результате в организме усиливается синтез NH3, который играет роль
«оберегающего компонента», сберегая Na+, K+:
Глутамин + Н2О→ глутаминовая кислота + NH3.
Причина кетозов – дефицит углеводов. При недостатке углеводов усиливается липолиз ТАГ в адипоцитах и в кровь поступают СЖК, 30%
которых захватывает печень. Повышенное количество жирных кислот усиливает кетогенез. Различают непатологические и патологические кетозы.
Непатологические кетозы возникают при углеводном голодании и при усиленной физической нагрузке.
Патологические кетозы возникают при тяжёлых формах сахарного диабета,
так как снижается поступление глюкозы в органы и ткани.
Гиперинсулинизм приводит к тканевому углеводному голоданию и кетозу.
При дефиците глюкокортикоидов, например, кортизола (стероидный диабет),
снижается скорость глюконеогенеза и возрастает чувствительность клеток к инсулину, что может привести к гипогликемии и кетозу.
При мочекислом диатезе у детей отмечается гиперурикемия (повышение уровня мочевой кислоты), которая сопровождается гипокалиемией,
кетонемией и кетонурией. Этот эффект можно объяснить тем, что мочевая кислота в адипоцитах ингибирует аденилатциклазу и нарушает передачу гормонального сигнала глюкагона и, как следствие, развивается гипогликемия и кетоз.