Материал: Застосування програмного забезпечення для рентгеноструктурного аналізу
. Виберіть диск, на якому знаходиться файл. 2. Виберіть папку, в якій зберігається файл. 3. Клацніть на назві файла два рази або виділіть його і натисніть клавішу Відкрити. У результаті буде завантажена експериментальна дифрактограма (рис. 3).
У цьому разі як приклад розглянуто аналіз магнієвого сплаву.
Сплави на основі магнію (MgAl) дуже широко використовуються як конструкційні
матеріали в машинобудівній та авіаційній промисловості завдяки їх легкості та
міцності. Зйомка даного зразка проводилася на дифрактометрі ДРОН-УМ1 з
фокусуванням по Бреггу-Брентано у монохроматичному мідному випромінюванні при
30 кВ, 30 мА. Запис дифрактограми здійснювали в цифровому ASCII форматі (x_y).
Час експозиції в точці вибраний 2 с, інтервал 2θ: 10-88°, крок сканування
0,05°.
Рисунок 3 - Вікно PCW 2.4 після завантаження експериментальної рентгенограми
Після завантаження експериментальної дифрактограми необхідно перевірити в програмі налаштування параметрів експерименту, які автоматично запам’ятовуються в файлі конфігурації (pcw.cfg) після попереднього нормального завершення роботи програми. Для цього вибираємо вкладку Diffraktion та натискаємо Experiment (рис. 4). У робочому вікні, що з’явилося, можна налаштувати такі параметри:
· тип джерела випромінювання;
· довжину хвилі Kα1 і Kα2 з урахуванням аномальної дисперсії;
· геометрію експерименту, щілини постійної чи змінної ширини;
· інтервал значень 2θ і початкове значення величини фону;
· максимальну ширину дифракційного профілю в одиницях FWHM.
Рисунок 4 - Налаштування параметрів експерименту
Для рентгенівського випромінювання можна використовувати такі матеріали аноду: Cr, Mn, Fe, Co, Ni, Cu, Zr, Nb, Mo, Ag і W. Тільки для цих випромінювань відповідні поправки (глибина проникнення, масовий коефіцієнт поглинання) враховуються при розрахунку порошкової дифрактограми. Це важливо, наприклад, для кількісного фазового аналізу. PowderCell розраховує дифракцію в геометрії Брегга-Брентано (Bragg-Brentano) і Гіне (Guinier), також враховується наявність чи відсутність монохроматора. Розрахунок можна робити і для дифракції нейтронів.
Зробивши попереднє припущення, що даний зразок містить
кристалічну структуру типу Мg, ми переходимо до створення теоретичної
дифрактограми структури типу Мg. Для створення нової структури необхідно
натиснути на панелі меню File та вибрати закладку New. У діалоговому вікні, що
з’явилося, потрібно ввести назву речовини, порядковий номер елемента за
таблицею Менделєєва, номер просторової групи, параметри елементарної комірки,
координати атомів в елементарній комірці, коефіцієнт заповнення позиції та
значення температурного фактора. Всі ці дані знаходимо, провівши огляд
літератури за даним типом кристалічної структури або скориставшись базою кристалоструктурних
даних (наприклад [3]). Дане вікно після заповнення необхідних параметрів матиме
вигляд вказаний на (рис. 5).
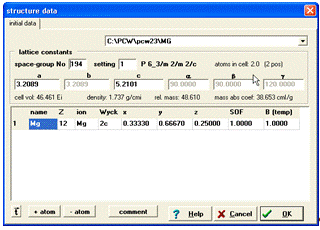
Рисунок 5 - Вікно
після введення атомних параметрів кристалічної структури Mg
Автоматично програма вираховує об’єм елементарної комірки,
щільність речовини, будує елементарну комірку і т. д. Для перегляду
елементарної комірки після створення теоретичної рентгенограми потрібно на
панелі меню натиснути Structure та вибрати підпункт Representation ON, елементарна
кристалічна ґратка магнію виглядатиме як на рис. 6.
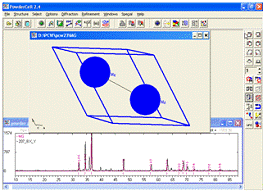
Рисунок 6 -
Розрахований дифракційний спектр та елементарна комірка гексагональної
структури магнію
Після створення нового файла кристалічної структури необхідно його зберегти в форматі PowderCell з розширенням (cel). Для цього необхідно натиснути на панелі меню File та вибрати закладку Save. В діалоговому вікні, що з’явилося потрібно ввести ім’я файла (наприклад Mg), під яким хочемо зберегти структурні дані. Іншим разом можна буде скористатися цим файлом для введення в програму структури Mg. Таким чином, можна створювати власну кристалоструктурну базу даних.
Створивши теоретичну дифрактограму, далі необхідно порівняти
її з експериментальною. Для цього розгортаємо на весь екран вікно з
дифрактограмами (рис. 7).
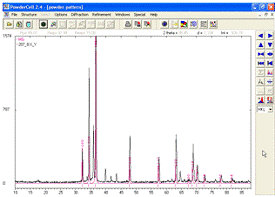
Рисунок 7 -
Експериментальна дифрактограма сплаву MgAl та розрахований дифракційний спектр
гексагональної структури магнію
Тепер у робочому вікні програми зображені тільки експериментальна та теоретична рентгенограми, накладені одна на одну. Як видно з рисунка, всі піки теоретичної дифрактограми збігаються з експериментальними, отже, дана фаза присутня в зразку.
Але на основі того, що експериментальна дифрактограма ще
містить піки, які відсутні в теоретичній, можна зробити висновок, що
досліджуваний матеріал складається з більшої кількості фаз. Оскільки
досліджуваний зразок містить у своєму складі Al, то вивчивши подвійну діаграму
стану Mg-Al [4], робимо припущення про те, що наступною кристалічною структурою
може бути Al12Mg17 (структура типу α - Mn). Тому створюємо
структурний файл даних для розрахунку теоретичної дифрактограми кубічної
структури Al12Mg17 аналогічно до того, як створювали відповідний структурний файл
для структури Mg. Дана кристалічна структура являється складнішою порівняно з
Mg, оскільки містить в своєму складі більшу кількість атомів (58) і відповідно
робоче вікно зі створення структурного файла даних для розрахунку теоретичної
дифрактограми виглядає таким чином (рис. 8).
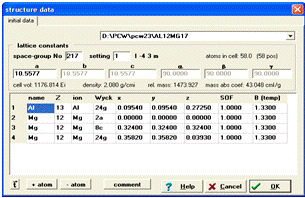
Рисунок 8 - Вікно
введення атомних параметрів кристалічної структури Al12Mg17
Створивши та зберігши структурний файл даних для розрахунку теоретичної рентгенограми Al12Mg17, можна подивитися на елементарні кристалічні ґратки обох фаз (рис. 9).
Закривши вікна з елементарними ґратками, бачимо дві
теоретичні дифрактограми, піки яких збігаються з максимумами на
експериментальній картині дифракції від сплаву MgAl (рис. 10).
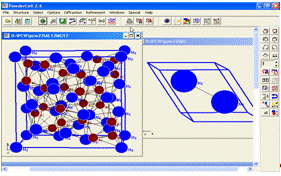
Рисунок 9 -
Елементарна комірка кубічної структури Al12Mg17 та гексагональної структури Mg
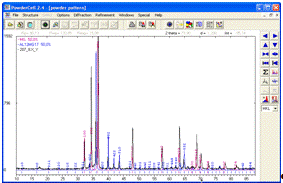
Рисунок 10 -
Експериментальна дифрактограма сплаву MgAl та розраховані дифракційні спектри
структур Mg та Al12Mg17
Відповідно з цим можна зробити висновок, що фазовий склад даного зразка ідентифікується як фази на основі Mg та Al12Mg17.
Провівши якісний аналіз шляхом порівняння теоретичних дифрактограм відповідних структур, що мають такі самі рентгенівські піки, як на дифрактограмах досліджуваного зразка, можна переходити до кількісного аналізу, тобто до розрахунку вагових концентрацій кожної з фаз.
Кількісний аналіз полягає в підборі параметрів кристалічних ґраток та інтенсивностей піків кожної з фаз. Для початку потрібно задати параметри для розрахунку профілю окремого рефлексу. Результуюча крива інтенсивності є згорткою функції профілю з штрих діаграмою. Щоб задати дані параметри потрібно вибрати на панелі меню пункт Diffraktion та підменю Phase options. У вікні, що з’явилося, задається група параметрів функції профілю окремого дифракційного піку (рис. 11).
powdercell полікристалічний меню diffraktion
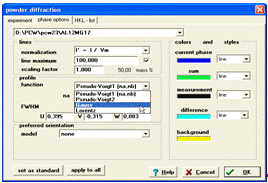
Рисунок 11 - Вікно
програми для введення параметрів функції профілю окремого дифракційного піку
Можуть бути задані такі профілі функції: Pseudo-Voigt1 (na,
nb) - Псевдо-Войт 1, Pseudo-Voigt2 - Псевдо-Войт 2, Lorentz - Лоренц, Gauss -
Гаусс. Також у даному робочому вікні задається ширина на піввисоті Full-width
at half-maximum (FWHM), даний параметр може бути постійним FWHM = const, бути
функцією від кута = FWHM = f(q) = const×tanqx, чи залежати від функції
FWHM = = f(U,V,W) = U×tan2q + V×tanq + W (функція Kагліоті [5]). В останньому випадку мають бути
введені значення U, V, W (рис. 12).
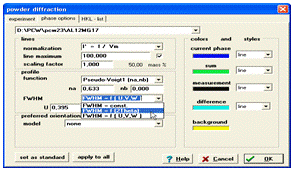
Рисунок 12 - Профільні параметри дифракційного максимуму
Параметр Preferred orientation у даному робочому вікні означає - переважна орієнтаціє (текстура). По замовчуванню - none (відсутність текстури), але при необхідності її можна задати моделлю Rietveld-Toraya (plate) - пластинчаста текстура, Rietveld-Toraya (needle) - голчаста текстура, March-Dollase - текстурна модель Марча-Долласа [6, 7].
На початковому етапі кількісного аналізу в програмі
PowderСell можна зафіксувати (зняти галочку) такі значення, як параметри
кристалічної гратки, параметри профілю функції U,V,W, na, nb. Щоб зафіксувати
дані значення потрібно вибрати на панелі меню пункт Refinement та підменю
Parameters і зняти галочки з необхідних місць (рис. 13).
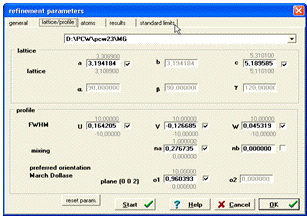
Рисунок 13 - Вікно
програми для введення параметрів кристалічної гратки, коефіцієнтів профільної функції
- (U,V,W) та текстури - (о1)
Охарактеризовується також (меню Refinement підменю general) степінь полінома, яким апроксимується лінія фону експериментально отриманої дифрактограми і число циклів уточнення, яке по замовчуванню дорівнює трьом, зміщення нуля детектора, зміщення зразка, коефіцієнт шкали та температурний фактор (рис. 14).
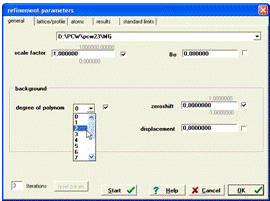
Рисунок 14 - Вікно
програми для введення шкального коефіцієнта, температурного фактора, степеня
полінома фону, числа циклів уточнення, зміщення нуля детектора та зсуву зразка
з осі гоніометра
Після закінчення налаштувань, задача полягає в підборі параметрів кристалічних ґраток та коефіцієнтів U,V,W, na, (nb=0). У результаті підбору цих параметрів різниця між інтенсивностями експериментальної і теоретичної дифрактограм повинні суттєво зменшитися.
Більш детальне уточнення і розрахунок кількості фаз
виконуються автоматично програмою при натисненні на панелі меню пункту
Refinement та підменю Start (рис. 15). Кількісні параметри після даного уточнення можна побачити в підменю Results
(рис. 16). Процедура уточнення може повторюватися декілька раз, для досягнення
максимальної відповідності теоретичної і експериментальної дифрактограм, для
цього декілька разів натискаємо підменю Start.
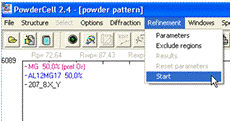
Рисунок 15 - Вибір
і запуск процедури уточнення названих параметрів
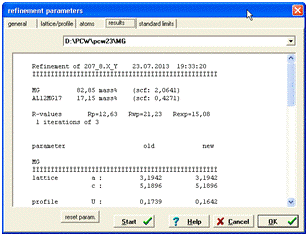
Рисунок 16 -
Параметри кількісного аналізу фазового складу сплаву MgAl після уточнення
Для візуального спостереження різниці між теоретичним і
експериментальним спектром у програмі Powdercell існує опція Show difference
(показати різницю), яка активується на правій боковій панелі інструментів (рис.
17).
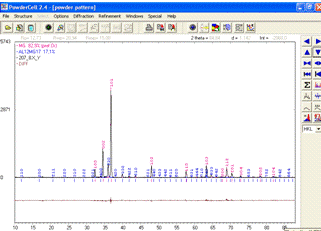
Рисунок 17 -
Результуючий вигляд повнопрофільного аналізу дифракційної картини з різницевим
спектром
Збіг експериментальної і розрахованої дифракційних картин математично оцінюється за значеннями трьох R-факторів: Rр - всієї картини; Rwp - з урахуванням інтенсивності (ваги) окремих піків та Rexp - очікуваного, виходячи зі статистики експерименту.
Після кожного етапу уточнення, у підменю Results, можна спостерігати кількісне співвідношення фаз, значення R - факторів, коефіцієнти полінома фону, періоди та кути елементарних комірок, індекси відбиття рефлексів, параметри півширини рефлексів.
Для даного випадку, після трьох циклів уточнення, маємо накладені одна на одну експериментальну та теоретичні дифрактограми. Цей зразок являється двофазним і елементарна комірка Al12Mg17 є досить складною, бо містить 58 атомів (як наслідок можливе перекриття рефлексів) За фазовим складом зразок містить 82,8 мас.% фази Mg з текстурою в напрямку 002 та періодами ґраток a = 3,194 Å (0,3194 нм), c = 5,189 Å (0,5189 нм), і 17,2 мас. % фази Al12Mg17 без текстури та періодом ґратки a = 10,580 Å (1,0580 нм).
Для збереження розшифрованої рентгенограми необхідно вибрати
на панелі меню Diffraction та підменю Copy to clipboard (рис. 18). Скопійовану
рентгенограму варто вставити в окремий текстовий документ і поряд розмістити
текст із параметрами, що знаходяться в підменю Results.
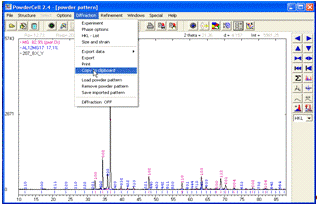
Рисунок 18 -
Копіювання в буфер розшифрованої рентгенограми
Результат кількісного фазового аналізу отримано в масових
частках, проте в деяких випадках доречно мати кінцевий результат в об’ємних
долях кожної з фаз. За допомогою PowderCell є можливість проводити розрахунок
кількісного аналізу трьома способами (рис. 19).
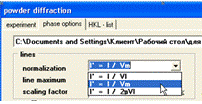
Рисунок 19 - Вибір
типу розрахунку частки фракції в суміші
Налаштування відповідних параметрів відбувається в меню
Diffraktion, підменю Phase options. Перший спосіб - знаходження вмісту у
об'ємних відсотках для кожної фази шляхом нормування інтенсивності I на квадрат
об’єму елементарної комірки (I / V2). Другий спосіб - розрахунок вмісту у
масових відсотках для кожної з фаз нормуванням інтенсивності I на об’єм та масу
їх елементарної комірки (I / VM). Ці два способи придатні для кількісного
розрахунку сумішей полікристалічних складових. Однак необхідно зауважити, що
внаслідок ефекту мікропоглинання, отримані результати будуть коректними тільки
у випадку близькості значень лінійного коефіцієнта поглинання рентгенівських
променів у наявних фазових складових та геометричної співрозмірності частинок
різних фаз. Третій спосіб - включає нормування інтенсивності I на лінійний
коефіцієнт поглинання рентгенівських променів (µ) та квадрат об’єму
елементарної комірки фази (I / 2µ V2), що дає результат в об'ємних частках для
кожної фази. Така нормалізація підтримує порівняння дифракційних картин тільки
для чистих одиночних фаз.
ВИСНОВКИ
У сучасних апаратах для рентгеноструктурних досліджень комп’ютер відіграє роль не тільки реєстратора інформації, але й управляє роботою всього приладу в цілому, а при наявності відповідних програм дозволяє обробляти отримані дифрактограми. Програмний комплекс PowderCell 2.4 широко використовується для повнопрофільного аналізу дифракційних картин. Використання цієї програми значною мірою звільнює дослідника від трудомістких «ручних» обчислень і зменшує похибку отриманих результатів.
Список використаної літератури
1. Rietveld, H. M. A profile refinement method for nuclear and magnetic structures. J. Appl. Cryst. 2 (1969), p. 65-71.
2. Powder Cell <http://www.ccp14.ac.uk/tutorial/powdcell/>
. Crystallography Open Database (COD) - <http://www.crystallography.net/>.
4. Диаграммы состояния двойных металлических систем: Д44 Справочник : в 3 т. / под общ. ред. Н. П. Лякишева. - М.: Машиностроение, 1996. - 992 с. ил.
5. Caglioti G., Paoletti A. & Ricci, F. P. : Choice of collimators for a crystal spectrometer for neutron diffraction. Nucl. Instrum. 3 (1958), p. 223-228.
6. March, A.: Mathematische Theorie der Regelung nach der Korngestalt bei affiner Deformation. Z. Kristallogr. 81(1932), 285-297.
7. Dollase, W.A.: Correction of intensities for preferred orientation of the March model. J.Appl.Cryst. 19(1986), 267-272.
. Уманский Я. С. Кристаллография, рентгенография и электронная микроскопия / Я. С. Уманский, Ю. А. Скаков, А. Н. Иванов, Л. Н. Расторгуев. - М. : Металлургия, 1982. - 632 с.
. Теоретический расчет рентгенограммы поликристалла: Описание лабораторной работы по курсу «Рентгеноструктурный анализ» / сост. : Т. В. Панова, В. И. Блинов. - Омск : Омск.гос. ун-т, 2004. - 20 с.