Материал: Общая фармакология
2. Распределение
При попадании в общий кровоток липофильные неполярные вещества распределяются в организме относительно равномерно, а гидрофильные полярные вещества — неравномерно, Препятствиями для распределения гидрофильных полярных веществ являются, в частности, гисто-гемагпические барьеры, т.е. барьеры, отделяющие некоторые ткани от крови. К таким барьерам относятся гематоэн-цефалический, гематоофтальмический и плацентарный барьеры.
Гематоэнцефалический барьер образован слоем эндотелиальных клеток капилляров мозга, в котором отсутствуют межклеточные промежутки. Гематоэнцефалический барьер препятствует проникновению гидрофильных полярных веществ из крови в ткани мозга. При воспалении мозговых оболочек проницаемость гематоэнцефа-лического барьера повышается.
Гематоофтальмический барьер препятствует проникновению гидрофильных полярных веществ из крови в ткани глаз.
Плацентарный барьер во время беременности препятствует проникновению ряда веществ из организма матери в организм плода.
Для характеристики распределения лекарственного вещества используют кажущийся объем распределения - Vd (Volume of distribution).
В
системе однокамерной фармакокинетической
модели![]() ,
,
где D — доза, Со - начальная концентрация. Поэтому кажущийся объем распределения можно определить как гипотетический объем жидкостей организма, в котором после внутривенного введения, при условии мгновенного и равномерного распределения концентрация вещества равна его концентрации в плазме крови. Vd определяют в литрах или л/кг.
Если для условного человека с массой тела 70 кг Vd = 3 л (объем плазмы крови ), это означает, что вещество находится в плазме крови, не проникает в форменные элементы крови и не выходит за пределы кровеносного русла.
Vd = 15 л означает, что вещество находится в плазме крови (3 л), в межклеточной жидкости (12 л) и не проникает в клетки тканей.
Vd = 40 л (общее количество жидкости в организме) означает, что вещество распределено во внеклеточной и внутриклеточной жидкости.
Vd = 400 - 600 -1000 л означает, что вещество депонировано в периферических тканях и его концентрация в крови низкая. Например, для имипрамина (трициклический антидепрессант) Vd = 23 л/кг, т.е. примерно 1600 л. В связи с этим концентрация имипрамина в крови очень низкая и при отравлении имипрамином гемодиализ не эффективен.
3. Депонирование
При распределении лекарственного вещества в организме часть вещества может задерживаться (депонироваться) в различных тканях. Из «депо» вещество высвобождается в кровь и оказывает фармакологическое действие. Липофильные вещества могут депонироваться в жировой ткани. Так, средство для внутривенного наркоза тиопентал-натрий вызывает наркоз, который продолжается 15—20 мин. Кратковременность действия связана с тем, что 90% тиопентала-натрия депонируется в жировой ткани. После прекращения наркоза наступает посленаркозный сон, который продолжается 2—3 ч и связан с действием препарата, высвобождаемого из жирового депо.
Антибиотики из группы тетрациклинов на длительное время депонируются в костной ткани. Тетрациклины не рекомендуют назначать детям до 8 лет, так как, депонируясь в костной ткани, они могут нарушать развитие скелета.
Многие вещества депонируются в крови, связываясь с белками плазмы крови. В соединении с белками плазмы вещества не проявляют фармакологической активности. Однако часть вещества высвобождается из связи с белками и оказывает фармакологическое действие. Вещества, которые более прочно связываются с белками, могут вытеснять вещества с меньшей прочностью связывания. Действие вытесненного вещества при этом усиливается, так как увеличивается концентрация в плазме крови его свободной (активной) формы. Например, сульфаниламиды, салицилаты могут таким образом усиливать действие назначаемых одновременно непрямых антикоагулянтов. При этом свертываемость крови может чрезмерно снижаться, что ведет к кровотечениям.
4. Биотрансформация
Большинство лекарственных веществ в организме подвергается превращениям (биотрансформации). Различают метаболическую трансформацию (окисление, восстановление, гидролиз) и конъюгацию (ацетилирование, метилирование, образование соединений с глюкуроновой кислотой и др.). Соответственно, продукты превращений называют метаболитами и конъюгатами. Обычно вещество подвергается сначала метаболической трансформации, а затем конъюгации. Метаболиты, как правило, менее активны, чем исходные соединения, но иногда оказываются активнее (токсичнее) исходных веществ. Конъюгаты обычно малоактивны.
Большинство лекарственных веществ подвергается биотрансформации в печени под влиянием ферментов, локализованных в эндоплазматическом ретикулуме клеток печени и называемых микросомальными ферментами (в основном изоферменты цитохрома Р-450).
Эти ферменты действуют на липофильные неполярные вещества, превращая их в гидрофильные полярные соединения, которые легче выводятся из организма. Активность микросомальных ферментов зависит от пола, возраста, заболеваний печени, действия некоторых лекарственных средств.
Так, у мужчин активность микросомальных ферментов несколько выше, чем у женщин (синтез этих ферментов стимулируется мужскими половыми гормонами). Поэтому мужчины более устойчивы к действию многих фармакологических веществ.
У новорожденных система микросомальных ферментов несовершенна, поэтому ряд лекарственных веществ (например, хлорамфеникол) в первые недели жизни назначать не рекомендуют в связи с их выраженным токсическим действием.
Активность микросомальных ферментов печени снижается в пожилом возрасте, поэтому многие лекарственные препараты лицам старше 60 лет назначают в меньших дозах по сравнению с лицами среднего возраста.
При заболеваниях печени активность микросомальных ферментов может снижаться, замедляется биотрансформация лекарственных средств, усиливается и удлиняется их действие.
Известны лекарственные вещества, индуцирующие синтез микросомальных ферментов печени, например, фенобарбитал, гризеофульвин, рифампицин. Индукция синтеза микросомальных ферментов при применении указанных лекарственных веществ развивается постепенно (примерно в течение 2 нед). При одновременном назначении с ними других препаратов (например, глюкокортикоидов, противозачаточных средств для приема внутрь) действие последних может ослабляться.
Некоторые лекарственные вещества (циметидин, хлорамфени-кол и др.) снижают активность микросомальных ферментов печени и поэтому могут усиливать действие других препаратов.
5. Выведение (экскреция)
Большинство лекарственных веществ выводится из организма через почки в неизмененном виде или в виде продуктов биотрансформации. В почечные канальцы вещества могут поступать при фильтрации плазмы крови в почечных клубочках. Многие вещества секретируются в просвет проксимальных канальцев. Транспортные системы, которые обеспечивают эту секрецию, малоспецифичны, поэтому разные вещества могут конкурировать за связывание с транспортными системами. При этом одно вещество может задерживать секрецию другого вещества и таким образом задерживать его выведение из организма. Например, хинидин замедляет секрецию дигоксина, концентрация дигоксина в плазме крови повышается, возможно проявление токсического действия дигоксина (аритмии и др.).
Липофильные неполярные вещества в канальцах подвергаются обратному всасыванию (реабсорбции) путем пассивной диффузии. Гидрофильные полярные соединения мало реабсорбируются и выводятся почками.
Выведение (экскреция) слабых электролитов прямо пропорционально степени их ионизации (ионизированные соединения мало реабсорбируются). Поэтому для ускоренного выведения кислых соединений (например, производных барбитуровой кислоты, салицилатов) реакцию мочи следует изменять в щелочную сторону, а для выведения оснований — в кислую.
Кроме того, лекарственные вещества могут выделяться через желудочно-кишечный тракт (выделение с желчью), с секретами потовых, слюнных, бронхиальных и других желез. Летучие лекарственные вещества выделяются из организма через легкие с выдыхаемым воздухом.
У женщин в период кормления грудью лекарственные вещества могут выделяться молочными железами и с молоком попадать в организм ребенка. Поэтому кормящим матерям не следует назначать лекарства, которые могут неблагоприятно воздействовать на ребенка.
Биотрансформация и экскреция лекарственных веществ объединяются термином «элиминация». Для характеристики элиминации используют константу элиминации — ке1 (ке) и период полуэлиминации - t1/2.
Константа элиминации показывает, какая часть вещества элиминируется в единицу времени. Например, внутривенно введено вещество А в дозе 10 мг; ке1 = 0,1/ч. Через 1 ч в плазме крови останется 9 мг, через 2 ч - 8,1 мг.
Период полуэлиминации — t1/2 — время, за которое концентрация вещества в плазме крови снижается наполовину. В основное время элиминации t1/2 не зависит от дозы вещества и одинаков в разное

Общий (total) клиренс определяется по формуле Clt = Vd * ke[.
Другими словами, Clt показывает, какая часть объема распределения освобождается от вещества в единицу времени.
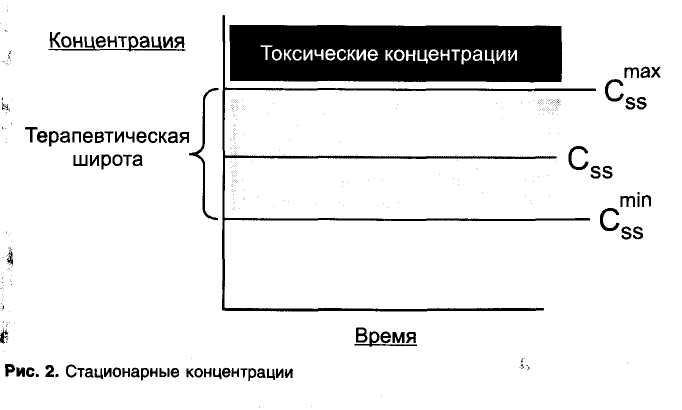
Для оптимального терапевтического эффекта и для предупреждения токсического действия необходимо поддерживать в плазме крови постоянную (стационарную) терапевтическую концентрацию лекарственного вещества. Стационарную концентрацию обозначают как Css (steady-state concentration). В справочниках и руководствах по фармакологии приводят значения средних терапевтических концентраций для наиболее употребительных лекарственных веществ.
Определяют также минимальную терапевтическую концентрацию (минимальную эффективную концентрацию) — Cssmin и максимальную терапевтическую концентрацию (максимальную безопасную концентрацию) - Cssmax, выше которой концентрации становятся токсическими. Интервал между Cssmin и Cssmax соответствует терапевтической широте (рис. 2). Чем больше терапевтическая широта лекарственного средства, тем легче его использовать в практической медицине. Наоборот, при малой терапевтической широте увеличивается вероятность попадания в зону токсических концентраций.
Для поддержания средней терапевтической концентрации лекарственного вещества можно вводить раствор этого вещества внутривенно капельно. При этом концентрация вещества в плазме крови сначала повышается быстро, затем медленнее и, наконец, устанавливается стационарная концентрация, при которой скорость введения вещества равна скорости его элиминации (биотрансформация + экскреция). Скорость введения определяют по формуле

Однако значительно чаще лекарственные вещества назначают внутрь или в виде отдельных инъекций. В этих случаях целесообразно сначала вводить нагрузочную дозу для быстрого достижения терапевтической концентрации, а затем назначать малые дозы, которые поддерживают терапевтическую концентрацию, — поддерживающие дозы.
Б. Фармакодинамика
Фармакодинамика - фармакологические эффекты, механизмы действия, локализация действия, виды действия лекарственных веществ.
Фармакологические эффекты лекарственного вещества — изменения в деятельности органов, систем организма, которые вызывает данное вещество (например, усиление сокращений сердца, снижение артериального давления, стимуляция умственной деятельности, устранение страха и напряженности и т.п.). Как правило, каждое вещество вызывает ряд характерных для него фармакологических эффектов. В каждом конкретном случае используют лишь определенные эффекты лекарственного средства, которые определяют как основные эффекты. Остальные (не используемые, нежелательные) фармакологические эффекты называют побочными эффектами.
Механизмы действия лекарственных веществ — способы, которыми вещества вызывают фармакологические эффекты. К основным вариантам механизмов действия относятся действие на: 1) специфические рецепторы, 2) ферменты, 3) ионные каналы, 4) транспортные системы.
Большинство лекарственных веществ действует на специфические рецепторы. Эти рецепторы представлены чаще всего функционально активными белковыми молекулами; взаимодействие с ними дает начало биохимическим реакциям, которые ведут к возникновению фармакологических эффектов.
Различают специфические рецепторы, связанные с клеточными мембранами (мембранные рецепторы), и внутриклеточные рецепторы.
Мембранные рецепторы делят на: 1) рецепторы, сопряженные с ионными каналами, 2) рецепторы, сопряженные с ферментами, 3) рецепторы, взаимодействующие с G-белками.
К рецепторам, сопряженным с ионными каналами, относятся, в частности, N-холинорецепторы и ГАМКА -рецепторы.
При стимуляции N-холинорецепторов (никотиночувствительные холинорецепторы) открываются сопряженные с ними натриевые каналы. Вход ионов Na+ в клетку обусловливает деполяризацию клеточной мембраны и возбудительный эффект.
ГАМКА -рецепторы непосредственно сопряжены с хлорными каналами. Стимуляция ГАМКА-рецепторов ведет к открытию Сl--каналов, входу ионов Сl-, гиперполяризации клеточной мембраны и тормозному эффекту.
К рецепторам, которые сопряжены с ферментами, относятся, в частности, рецепторы инсулина, сопряженные с тирозинкиназой.
Рецепторы, взаимодействующие с G-белками, — М-холинорецепторы (мускариночувствительные холинорецепторы), адренорецепторы, дофаминовые рецепторы, опиоидные рецепторы и др.
G-белки, т.е. ГТФ-связывающие белки, локализованы в клеточной мембране и состоят из α-β-γ-,субъединиц. При взаимодействии лекарственного вещества с рецептором α -субъединица G-белка соединяется с ГТФ (GTP) и воздействует на ферменты или ионные . каналы. Один рецептор взаимодействует с несколькими G-белками, а каждый комплекс а-субъединицы G-белка с ГТФ действует ;на несколько молекул фермента или на несколько ионных каналов. Таким образом осуществляется механизм амплифайера (усилителя): при активации одного рецептора изменяется активность многих молекул фермента или многих ионных каналов.
Одними из первых были обнаружены G-белки, связанные с β 1-адренорецепторами сердца. При активации симпатической иннервации сердца возбуждаются β 1-адренорецепторы; через посредство G-белков активируется аденилатциклаза; из АТФ образуется цАМФ, активируется протеинкиназа, при действии которой фосфорилиру-ются и открываются кальциевые каналы.
Увеличение входа ионов Са2+ в клетки синоатриального узла ускоряет 4-ю фазу потенциала действия — сокращения сердца учащаются. Открытие Са2+-каналов в волокнах рабочего миокарда ведет к увеличению концентрации Са2+ в цитоплазме (вход Са2+ способствует высвобождению Са2+ из саркоплазматического ретикулума). Ионы Са2+ связываются с тропонином С (составная часть тропонин-тро-помиозина); таким образом уменьшается тормозное влияние тропонин-тропомиозина на взаимодействие актина и миозина - сокращения сердца усиливаются (рис. 3).
При активации парасимпатической иннервации сердца (блуждающие нервы) возбуждаются М2-холинорецепторы и через посредство G-белков аденилатциклаза угнетается — сокращения сердца урежаются и ослабляются (в основном ослабляются сокращения предсердий, так как парасимпатическая иннервация желудочков относительно бедна).
Таким образом, G-белки могут оказывать на аденилатциклазу как стимулирующее, так и угнетающее влияние. Стимулирующие G-белки обозначили как Gs -белки (stimulate), а угнетающие — Gi-белки (inhibit) (рис. 4).
При возбуждении М1-холинорецепторов, М3-холинорецепторов, α1-адренорецепторов через Gq белки активируется фосфолипаза С, которая способствует тому, что из фосфатидилинозитол-4,5-дифос-фата образуются инозитол-1,4,5-трифосфат и диацилглицерол. Ино-зитол-1,4,5-трифосфат стимулирует высвобождение ионов Са2+ из саркоплазматического ретикулума (рис. 5, 6).