Материал: Газова хроматографія в аналізі повітря
Проте, погрішність виміру, складова
не більше 5 (відн.), досягається практично на будь-якому сучасному газовому
хроматографі. У спеціальних експертизах можливо проводити хроматографічні
аналізи з погрішністю 1-2.
.1.4 Основні блоки газового хроматографа
Прилади, за допомогою яких виконується колонкове хроматографічне розділення сумішей і їх аналіз, називаються хроматографами. Хроматографи - прилади або установки для хроматографічного розділення і аналізу сумішей речовин.
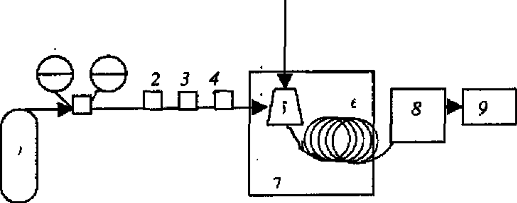
Рисунок1.1 - Схема газового
хроматографа: 1 - балон зі стисненим газом; 2 - регулятор витрати газу;
3-вимірювач витрати газу; 4 - фільтр; 5 - дозатор-випарник; 6 -хроматографічна
колонка; 7 - термостат; 8 - детектор; 9 - пастка.
Інертний газ-носій (рухома фаза) з балона 1 через регулятор тиску 2, вимірювач витрат (швидкості подачі) газу 3 та фільтр 4 потрапляє в дозатор-випарник (інжектор) 5, куди вводиться за допомогою шприца рідка або газоподібна проба. У дозаторі-випарнику створюється така температура, за якої компонента проби, що аналізується, перебувають в газоподібному або пароподібному стані. Далі під дією газу-носія компоненти проби переносяться у хроматографічну холонку 6, яка знаходиться в термостаті 7 і безперервно "промивається" потоком газу-носія з оптимальною швидкістю. Після розділення в колонці компоненти проби потрапляють у детектор 8. Усіма блоками сучасного хроматографа керує спеціальне програмне забезпечення, встановлене у комп’ютері. На виході з хроматографічної колонки іноді прилаштовують поглинач газів -“пастку” 9[5].
Конструкція газових хроматографів забезпечує етапу швидкість подачі газу- носія та сталу або змінну за певною програмою температуру колонки. Для виконання газо-хроматографічного аналізу хроматограф спочатку виводять на сталий режим роботи, створенням сталої швидкості подачі газу носія в колонку, певної її температури, а також стабільності подачі необхідних газів у детектор. Потім за допомогою шприца вводять пробу. При цьому температура дозатора-випарника має забезпечувати практично миттєвий перехід проби з рідкого стану в пароподібний. При проходженні газу-носія крізь колонку компоненти проби у вигляді окремих зон потрапляють в елюат і на екрані комп'ютера реєструються відповідні криві елюювання. Якісний і кількісний аналіз виконують на основі кривих елюювання. Пробу можна вводити вручну за допомогою шприца у дозатор-випарник шляхом "проколу" гумової або іншої еластичної прокладки або пробовідбірником (автосамплером) - пристроєм, який сам відбирає і вводить пробу. Введення проби автосамплером забезпечує вищу ефективність (нерозмивання проби на вході в колонку) і автоматизацію аналізу. Дозатор (інжектор) - посудина з мембраною, з'єднана з початком колонки.
Кількість проби для аналізу зв’язана з продуктивністю і ефективністю колонки, а також з чутливістю детектора. Чим більша маса або об'єм проби, тим більший сигнал детектора і тим більша чутливість хроматографа, але дуже велика проба викликає перевантаження колонки, спотворення форми піків і зменшує ефективність розділення. Матеріал колонок має бути інертний до речовин, з яких складається проба. Найчастіше їх виготовляють з нержавіючої сталі, міді, алюмінію, латуні, скла, кварцу і тефлону. Хроматографічні колонки різних довжин і діаметрів (рис.1.2) виготовляють із матеріалів, які не взаємодіють з газом-носієм і компонентами проби. Зручною формою хроматографічної колонки є спіральна,бо дозволяє на компактніше розмістити її у термостаті.
Одним із найважливіших блоків хроматографічної колонки є детектор - прилад, функція якого полягає в безперервній фіксації залежності концентрації, потоку чи кількості речовини на виході з колонки від часу.
Диференційні - передають миттєво значення концентрації чи потоку в часі. Їх поділяють в свою чергу на:
а) потокові - фіксують добуток концентрації на швидкість потоку.
б) концентраційні - фіксують зміну концентрації на виході з колонки.
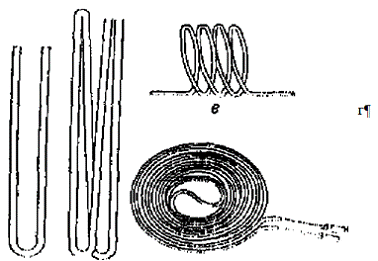
Рисунок 1.2 - Газохроматографічні колонки
а - U-подібна; б - W-подібна; в - спіральна
г - плоскоспіральна.
Інтегральні - сумують кількість речовини за певний проміжок часу, передають загальну кількість речовини, що пройшла через детектор.
Детектори поділяють також на універсальні і селективні. Універсальні дектори реєструють властивість, яку мають усі речовини, селективні - вимірюють властивість, притаманну певному виду молекул.
Кожний детектор характеризується такими основними величинами:
· чутливість:
· межа детектування
· інерційність
У газовій хроматографії використовуються такі основні принципи детектування:
) Залежність теплопровідності газової суміші від її складу (детектор за теплопровідністю).
) Тепловий ефект спалювання горючих компонентів на нагрітій до високої температури платиновій нитці (термохімічний детектор).
) Іонізація органічних сполук у полум'ї водневого пальника (полум'яно-іонізаційний детектор).
) Іонізація органічних сполук під дією зіткнення з метастабільними атомами аргону, які утворюються від впливу радіоактивного β-випромінювання (аргоновий детектор).
) Захоплення електронів молекулами органічних сполук (детектор за захопленням електронів).
) Зменшення іонізації полум'я з атомами лужних металів у присутності фосфор- і галогеновмісних сполук (термоіонний детектор).
) Залежність густини газової суміші від її складу (детектор за густиною).
) Специфічне випромінювання фосфор- і сірковмісних сполук у полум'ї водню (полум'яно-фотометричний детектор).
) Залежність зміни тиску на діафрагмі від складу газової суміші (діафрагмовий детектор).
Крім цих методів,
постійно розробляються нові принципи детектування для забезпечення більшої чутливості
аналізу. Найбільш розповсюдженими детекторами є детектори за теплопровідністю і
полум'яно-іонізаційні детектори.
2. Експериментальна частина
.1 Апаратура й матеріали
Газовий хроматограф « хромосом ГХ- 1000 », забезпечений іонізаційно - полум'яним детектором (поріг чутливості по пропану 1 х 10-11 г / с).
Мікрошпріцем типу " HAMILTON ": 10, 100, 1000 мм 3, похибка дозування 1 %.
Хроматографічна колонка 1:матеріал - скло; довжина 55-60 м; внутрішній діаметр 0,6 - 0,65 мм; нерухома фаза - поліетиленгліколь (карбовакс 20М ), ефективність колонки не менш 60000 еф. тт. по толуолу при 70 °C, виробництва НП ТДВ " Люкеп".
Хроматографічна колонка 2:матеріал - скло; довжина 55-60 м; внутрішній діаметр 0,6 - 0,65 мм; нерухома фаза - полідиметилсилоксан (SE 30 ), ефективність колонки не менше 50000 еф. тт. по толуолу при 70 °C, виробництва НП ТДВ "Люкеп".
Градуйовані скляні посудини(віали)
для приготування градуювальних проб ємністю 2 см3.Скляні посудини
для приготування градуювальних газоподібних проб ємністю 100-120 см3,
забезпечені закрученими пробками і еластичними прокладками (Supelco).
Електроаспіратори мод. 822 по ТУ 25-11.1660-85, наведена похибка ± 5 % (
ротаметр 0-20 л / хв).
.2 Реактиви та посуд
Калібрування віал проводили при температурі 20 ± 0,2 °С. Забезпечені мітками віали ретельно промивали, висушували і зважували на аналітичних вагах з точністю до 0,001 м. Потім заповнювали дистильованою водою до позначки «1 мл» і знову зважували. Істинний обсяг обчислювали за формулою
V = ( m1 - m2)
/ ρ (см3) (2.1)
де V - істинний обсяг віал, см3; 1 - маса заповненої Віали, г; 2 - масса порожньої віали, г;
ρ - щільність води при температурі зважування, г/см3.
Калібрування судин проводили при температурі 20 ± 0,2 °С. Скляні судини об'ємом приблизно 100 см3 ретельно промивали, висушували, вносили в нього перемішуючий елемент магнітної мішалки і зважували на аналітичних вагах з точністю до 0,1 м. Потім заповнювали дистильованою водою до верхнього зрізу горловини і знову зважували. Об'єм посудини обчислювали за формулою
= ( m1 - m2)
/ ρ (см3) (2.2)
де V - об'єм посудини, см3 ; 1 - маса заповненого судини, г; 2 - маса порожнього судини, г;
ρ - щільність води при температурі зважування, г/см3.В якості пробки використовували плоску гумову прокладку, розміщену всередині закрученої кришки і прилеглу до горловини посудини зверху.
Після повторної сушки внутрішню поверхню судини силанизували 10% розчином діметіл діхромсілану в толуолі.
2.3 Приготування градуювальних
газоподібних проб
Для відтворення одиниць концентрації визначених компонентів готували вихідний розчин визначених компонентів методом змішування чистих речовин з подальшим їх розведенням. При проведенні градуювання на колонці №1 (карбовакс 20М) як розчинник застосовували н- пропілацетат, при проведенні градуювання на колонці №2 (SE30) як розчинник застосовували н-пропанол. Допускається застосування іншого розчинника, пік якого на хроматограмі НЕ інтерферує з піками визначених компонентів.
Стандартний розчин 1. У калібровану віалу місткістю 2 см3, за допомогою мікро шприца на 50 мм 3 (проколюючи прокладку закученої пробки віали) вносили 15 - 20 мг кожного компонента, потім обсяг розчину у віалі доводили до мітки ( 1 см3) відповідним розчинником.
Стандартні розчини 2-6. Після ретельного перемішування, певний обсяг стандартного розчину 1 за допомогою микрошприца вносили в калібровану віалу ємністю 2 см3, потім обсяг розчину у віалі доводили до мітки (1 см3) відповідним розчинником.
Градуювальні суміші шкідливих речовин з повітрям. Після ретельного перемішування 30 мм3 стандартного розчину 2-6 за допомогою микрошприца поміщали на дно градуювальної посудини, помістили в посудину перемішуючий елемент, закрили його пробкою і помістили на увімкнену магнітну мішалку. Перемішування проводили протягом 40-60 хвилин. Масова концентрація Ci компонентів газоподібній градуювальній суміші дорівнює
i
= 1000 ( Кi х mi х Vsol ) / ( Vm х
V ) (2.3)
де Ci - концентрація компонента в градуювальній пробі, мг/м3; i - масса компоненту, взята для приготування вихідного розчину № 1, мг ; sol - об'єм розчину, що вводиться в скляну калібрувальну посудину, мм3 ; m - істинний обсяг стандартного розчину №1, см3;- об'єм скляної калібрувальної посудини, см3 ;
К1 - коефіцієнт розведення при приготуванні стандартного розчину № 2, рівний відношенню обсягу стандартного розчину № 1, поміщеного в градуювальну віалу до обʼєму градуювальної віали 2 (Vp-ну1/Vв2).
Кожну градуювальну газоподібну пробу хроматографували 5 разів, починаючи з самої низької концентрації визначених компонентів. Умови проведення градуювання наведені в таблиці 2.1
Відбір проби з калібрувального
посуду проводили шляхом проколювання гумової прокладки голкою газового шприца,
введення проби в газовий хроматограф виробляють тим же газовим шприцом.
.4 Визначення концентрації парів
летких органічних сполук при їх спільній присутності в газових викидах
промислових підприємств б методом капілярної газорідинної хроматографії
Запропоновано методику для визначення вмісту парів 1,4 - діоксану, акрилонітрилу, ацетону, бензолу, і - бутанолу, і - бутилацетату, ізопентілацетату, изопропанолу, п- ксилолу, м - ксилолу, о-ксилолу, кумолу, метанолу, метилетилкетону, н-бутанолу, н-бутилацетату, н-пентілацетату, н-пентану, н-гексану, н-гептану, н-октана, сольвента нафтового, псевдокумолу, стиролу, толуолу, уайт-спирту, трихлоретилену, етанолу, етилацетату, етилбензолу, етілцеллозольву в газових викидах промислових підприємств при спільній присутності за допомогою капілярної газорідинної хроматографії. [4]
При контролі повітря робочої зони промислових підприємств та їх газових викидів найчастіше доводиться мати справу з парами органічних розчинників. Виходячи з вищесказаного, вибрали 29 пріоритетних речовин, що виділяються фарбувальними камерами, і запропонували методику їх визначення у повітряних матрицях
Методика забезпечує вимірювання
вмісту аналізованих компонентів у промислових викидах з точністю близько 20% в
діапазоні концентрацій від 10 мг/м3 до 5000мг/м3 при відборі
проби об'ємом 100 - 500 см3.
.4.1 Результати методики
Градуювальну характеристику, виражену залежністю площ хромато-графічних піків визначених компонентів від концентрацій їх парів у градуювальній газоподібної пробі, встановлювали за 5 сумішами на кожній з двох колонок.
Застосування градуювальних скляних
посудин невеликого об'єму для приготування атестованих пара-повітряних сумішей
можливо за рахунок застосування розчинів низької концентрації. Діапазон
концентрацій, при якому зберігається лінійність градуювальної залежності,
залежить від температури кипіння аналіту. При температурах кипіння що не
перевищують 150 °С лінійність зберігається в діапазоні від 0 до 1000 мг/м3
для градуювальних судин об'ємом 120-140 см3. При інтенсивному
перемішуванні розчин повністю випаровується в закритій посудині протягом 30-40
хвилин (розчинник - н- пропілацетат, обсяг проби 10 мкл). В результаті
градуювання стандартне відхилення не перевищувало 8%, а коефіцієнт кореляції не
опускався нижче 0,99 для лінійної залежності типу y = ax за кожним компонентом.
Лінійна залежність зазначеного типу зберігалася у всьому діапазоні концентрацій
градуювальних розчинів.
Таблиця 2.1 - Умови проведення
градуювання
Показник
Значення
Обʼєм проби,мм3
300-500
Температура термостату колонки, ° С
70
Температура випарника, ° С
150
Температура детектору, ° С
150
Сумарна витрата газу-носія, мл/хв(на одну
колонку)
35
Коефіцієнт ділення потоку на вході в колонку
1/5:
1/5
Витрата газів для іонізаційно-полум'яного
детектора, мл/хв:
Водень
30
Повітря
300
піддування газу-носія в детектор
30
У випарник хроматографа вводили
300-500 мм3 проби. Для отримання кількісних результатів
використовували колонку, на якій виробляли градуювання хроматографа. Другу
колонку використовували для підтвердження ідентифікації компонентів. Вимірювали
площу хроматографічних піків, відповідних визначеним компонентам. Кожну пробу
хроматографіровалі не менше трьох разів.
Порядок елюювання компонентів:
На колонці 1 (карбовакс 20М): н-
пентан, н- гексан, н- гептан, н- октан, ацетон, етилацетат, метилетилкетон,
метанол, ізопропанол, танол, бензол, трихлоретилен, акрилонітрил, і -
бутилацетат, толуол, 1,4- діоксан, н- бутилацетат, і - бутанол,
ізопентілацетат, етилбензол, п- ксилол, м - ксилол, н- бутанол, кумол, о- ксилол,
н- пентілацетат, етілцеллозольв, стирол, псевдокумол ;
На колонці 2 (SE 30): метанол,
етанол, ацетон, ізопропанол, акрилонитрил, н- пентан, метилетилкетон,
етилацетат, н- гексан, і - бутанол, н- бутанол, бензол, 1,4 - діоксан,
трихлоретилен, етілцеллозольв, н- гептан, і - бутилацетат, толуол,
н-бутилацетат, н- октан, етилбензол, ізопентілацетат, н- пентілацетат, п-
ксилол, м -ксилол, стирол, о- ксилол, кумол, псевдокумол. [11]
При визначенні кількісного вмісту
парів вуглеводнів в промислових викидах, методом зовнішнього стандарту, для
цього використовували середнє арифметичне площ піків, отриманих при обробці не
менше 3х хроматограм. Концентрацію компонента в повітрі (C,мг/м3)
визначали з калібрувального графіка або за формулою. У разі, якщо концентрація компонента
в робочій пробі перевищує 1000 мг/м3, слід зменшити об'єм проби, що
вводиться в газовий хроматограф. тоді концентрацію компонента в повітрі
визначали за формулою
i
= Hi х Vg / ( a х Va ), см3 (2.4)
де Vg - об'єм проби, що
вводиться в хроматограф на стадії градуювання a -об'єм проби, що
вводиться в хроматограф на стадії аналізу, см3.
Рисунок 2.1 - Типова хроматограмма
суміші пари розчинників.
Колонка 1(CW 20M)
.5 Можливості визначення складу
складної газової суміші на хроматографі «ЦВЕТ - 800»
.5.1 Методика проведення аналізу
Метод кількісного газового аналізу
можна застосовувати тільки після попереднього встановлення для кожного
визначуваного компонента газової пробикількісної залежності між змістом цього компонента,
що вводиться в колонку, і площею його хроматографічного піку, який фіксує
детектор. Ця кількісна залежність може бути задана або градуювальним
коефіцієнтом пропорційності, або градуювальним графіком.
Рисунок 2.2 - Типова хроматограмма
суміші пари розчинників.
Колонка 2(SE 30)
У першому випадку готують атестовану
газову суміш із заданими концентраціями компонентів С'i (за
можливості близькими до їх передбачуваної концентрацій Сi впробі);готову
суміш хроматографують, вводячи в колонку певний об'єм V отриманої суміші;
визначають площі піків S'i і розраховують градуювальні коефіцієнти ki
= С'i /S'i; далі аналізують пробу, яку вводять в
хроматограф в тому ж об'ємі V; вимірюють площі піків S i і знаходять
концентрації компонентів упробі (в обʼємних відсотках) за формулою
С i = 100 kS i
У другому випадку готують серію
атестованих сумішей з різними заданими концентраціями компонентів; приготовані
суміші хроматографують, вводячи в колонку один і той же об'єм V кожної суміші;
визначають площі відповідних піків; для кожного компонента суміші будують
градуювальні графіки S'i = f(С' i); далі хроматографують
пробу об'ємом V, визначають площі Si відповідних піків і по графіках
знаходять концентрації компонентів Сi в пробі. Очевидно, що в
другому випадку досягається більш висока точність аналізу. Але для цього
необхідно мати великий набір атестованих газових сумішей, що містять усі
компоненти, передбачувані наявністю в пробі, причому в різних кількісних
співвідношеннях.
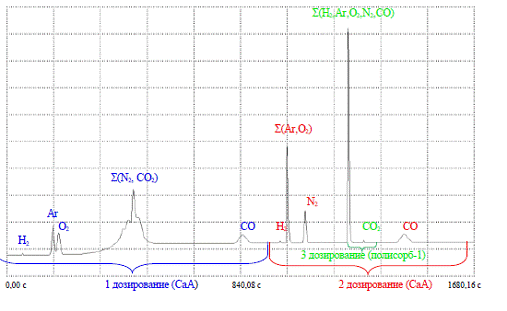
Хроматограф "Цвет-800" в
заводському виконанні був укомплектований тільки однією атестованою газовою
сумішшю з наступними концентраціями компонентів (в об'ємних відсотках): H2
- (0,81±0,08); О2 -(0,85±0,08); Ar-(1,05±0,10);N2-(1,00±0,10);
CO2-(1,20±0,12); CO- (0,060±0,006);
Передбачалося, що в пробах із
стендів EAGLE і АНГАРА будуть присутніми саме ці гази, причому розділення H2,
O2, Ar, N2 і CO проводитиметься на цеолітовому сорбенті
СаА -МС- 904 з фракціями 0,25-0,6 мм, а розділення СО 2 від інших
компонентів - на сорбенті типу полісорб- 1 з фракціями 0,25-0,5 мм.
.5.2 Результати аналізу
При відробітку методики проведення
якісного і кількісного газового аналізу на хроматографі "Цвет-800"
використовувалася вказана вище атестована газова суміш. Відробіток полягав у
виборі оптимальних режимів хроматографування цієї суміші з метою використання
вибраних режимів надалі при аналізі газових проб, відібраних з
експериментальних установок стендів EAGLE і АНГАРА.
При виборі типу колонки для
розділення в ній Ar і O2 були випробувані колонки діаметром 3 мм
різної довжини(3 і 6 м) з цеолітом і з'ясовані, що при температурах колонки і
+40, і 0 °С ці гази не розділяються через близькість часів їх утримання на
цеоліті при цих температурах та сильного розширення хроматографічних піків(сильного
розмивання газів по довжині колонки) незалежно від витрат гелію. Переходом на
колонку діаметром 2 мм і варіюванням температурою колонки і втрат газу-носія
встановлено, що повне разділення піків Ar і O2 в ізотермічному
режимі досягається при температурі -25 °С і витраті 20 см3/хв.
(збільшення і зменшення витрат гелію через колонку погіршувало якість
розділення цих компонентів).
Необхідно зауважити, що використання
колонки з цеолітом при температурі мінус 25 оС призводить до
небажано великого збільшення часів утримування N2 і СО, а також до
помітного збільшення (на ~20 хв.) часу десорбції N2 і, тобто, до
сильного розширення хроматографічного піку цього газу. Використання лінійного
програмування температури від - 25 до + 50 °С, скоротило час аналізу, але
положення піку N2 при цьому виявилося співпадаючим з фоновим піком
детектора ДТП (що реагує на підвищення температури колонки), що понизило
надійність обробки піку N2.
З урахуванням цього було з'ясовано,
що визначення концентрації N2 правильніше виконувати при другому
дозуванні суміші (другому введенні в газ-носій порції суміші об'ємом V) при
температурі колонки + 50 °С і витраті газу-носія 20 см3/хв.
Додатковою перевагою такого режиму є
можливість отримання піку CO2 при третьому дозуванні.
Для третього дозування, яке
проводилося через 180 с. після другого, використовувалася колонка з полісорбом
довжиною 3 м і діаметром 3 мм при температурі + 50 °С і витраті 30 см3/хв,
підключення до другого плеча ДТП(до першого плеча ДТП підключена колонка з
цеолітом). Це дозволило отримати на одній хроматограммі і сумарний пік H2+Ar+O2+N2+CO,
і окремо пік СО2.
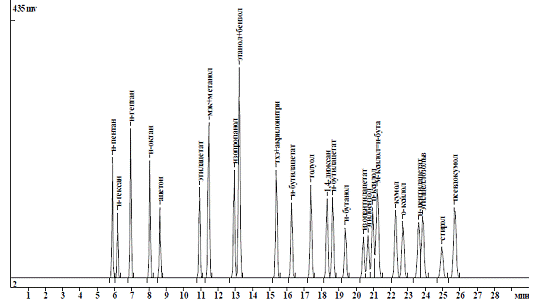
Рисунок 2.3 - Хроматограмма
атестованої суміші
Таким чином, було встановлено, що
оптимальні режими аналізу атестованої газової суміші повинні містити її
ізотермічні хроматографування після трьох дозувань, причому після першого
дозування визначаються тільки концентрації H2, Ar і O2
(колонки з цеолітом, температура - 25 °С, витрата 20 см3/хв), після
другого - тільки N2 і СО (колонка з цеолітом, температура При вивченні якісного і кількісного
складу газоподібних продуктів, що виникають при проведенні експериментів в
установках стендів EAGLE і АНГАРА, були проаналізовані 54 з 56 проб (таблиця
2.2), відібраних в ході виконання останніх одинадцяти експериментів.
Таблиця 2.2 - Кількість досліджених
газових проб
Стенд
Експеримент
Місце відбору (та кількість проб)
АНГАРА
IVR-2
ЭПП (3);
ОПР (3)
IVR-3
ЭПП (3);
ОПР (3)
IVR-3/1
ЭПП (3);
ОПР (3)
IVR-3/2
ЭПП (3);
ОПР (3)
UTD-М3
ЭПП (3);
ВЛ (2)
UTD-М2С
ЭПП (3);
ВЛ (2)
UTD-М5
ЭПП (3);
ВЛ (2)
EAGLE
PIDO
ЭПП (3);
ВЛ (2)
IDO-1
ЭПП (3);
ВЛ (2)
IDO-2
ЭПП (3);
ВЛ (2)
IDO-3
ЭПП (3);
ВЛ (2)
ЭПП - електроплавильна піч; ОПР -
облаштування прийому розплаву; ВЛ - верхня пастка.
Результати якісного аналізу
показали, що усі компоненти, що заздалегідь передбачалися, в сумішах дійсно є
присутніми в усіх відібраних газових пробах (тільки у пробах, відібраних в
експериментах UTD-М5, РIDO і IDO - 2, був відсутній кисень). Крім того, в усіх
пробах виявлялася присутність компонента, який можна було імовірно
ідентифікувати як метан.
Припущення вдалося підтвердити в
додаткових дослідженнях: при хроматографуванні спеціально придбаної атестованої
суміші,що містить, N2 з 1,05% (об.) CH4, було
встановлено, що час утримання CH4 точно відповідає часу утримування
виявленого в пробах компонента. (У цьому експерименті був також визначений
градуювальний коефіцієнт пропорціональності для метану).
Результати кількісного аналізу
дозволили простежити в кожному експерименті за змінами в часі концентрацій
компонентів в суміші, так як проби відбиралися з ЭПП, ОПР і ВЛ протягом 30 хв.
по три або по два рази в різні моменти часу: до зливу коріуму з ЭПП в ОПР або
ВЛ, відразу після зливу і приблизно через чверть години після зливу. З'ясовано,
що в пробах із стенду АНГАРА зміна концентрацій у часі були довільно різними, а
в усіх пробах із стенду EAGLE у цих змін спостерігалася наступна загальна
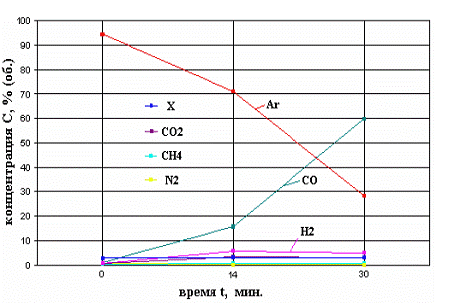
тенденція: значне підвищення концентрації СО (як приклад на Рис. 2.2.4
показаний характерний вид змін концентрації компонентів в газовій суміші в
ЭПП).
У окремих пробах, відібраних в
експериментах IDO - 1, IDO - 2 і IDO - 3, був присутній невідомий газовий
компонент Х, який доки не вдалося ідентифікувати.
Таким чином, результати проведення
якісного і кількісного аналізу складних газових сумішей свідчили про цілком
реальні можливості хроматографа "Цвет-800". Причому ці можливості
приладу можна помітним чином розширити, підвищивши при цьому точність
кількісного аналізу. [12]
Рисунок 2.4 - Зміни концентрацій
компонентів в газовій суміші, за певний час(хв.), що утворюється в ході
експерименту IDO - 1 на стенді EAGLE
2.6
Аналітичний огляд методів визначення мікрокількостей
акролеїну в повітрі
Для аналізу акролеїну
розроблені методи газової і рідинної хроматографії через високу летючість і
реакційну здатність акролеїну виникають проблеми з його уловлюванням з повітря
та зберіганням відібраних проб. Узагальнені відомості хроматографічних методів
визначення акролеїну з попередньою дериватизациею приведені в таблиці 2.3
Визначення акролеїну в
повітрі робочої зони методом ТШХ грунтовано на переводі акролеїну в нелетке
похідне з мета-фенілендіаміном і виявленні в ультрафіолетовому світлі на
хроматографічній пластинці у вигляді того, що флуоресценцює блакитним кольором плями.
Межа виміру 0,07 мг/м3 (об'єм проби 1,5 дм3). Відбір проб
проводиться із швидкістю 0,3 дм3/хв.. в поглинаючі прилади, що
містять 0,25%-й розчин гідроксиламіну гидрохлорида. Для проведення реакції
дериватизации реактивну суміш нагрівають на киплячій водяній бані 15 хв.
Отриманий дериват екстрагують 5 см3 хлороформу. Визначенню заважають
альдегіди, кетон, оксиди азоту. Велика кількість чутливих і селективних методів
аналізу альдегідів і кетонів грунтовані на їх взаємодії з
2,4-динітрофенілгидразином і аналізі 2,4-динітрофенілгідразо-, що утворюються
новий альдегідів(2,4-ДНФГ) хроматографічними методами в ультрафіолетовому
світлі в діапазоні довжин хвиль 350-380 нм. Цей метод ефективний для багатьох
альдегідів і кетонів. Проте при визначенні акролеїну і інших ненасичених
карбонілів виникають проблеми, що включають нестабільність 2,4-ДНФГакролеїну
впродовж відбору і зберігання проб, неповне хроматографічне ділення складної
суміші карбонілів, зазвичай присутніх в повітрі, великий час відбору проб з
низькими швидкостями потоку від 0,1 до1,0 л/хв при використанні картриджів
(звичайно 4-12 годин) для досягнення чутливості. [10]E. і співавтори при
порівняльному вивченні методів показали непридатність методу з 2,4-ДНФГ для
визначення акролеїну. Liu і співавтори дослідженнях за оцінкою способів
дериватизації шести альдегідів, у тому числі акролеїну, встановили, що втрати
2,4-ДНФГ акролеїну у відібраній пробі складають 32% через 24 години зберігання
за кімнатних умовах. При визначенні акролеїну і формальдегиду в повітрі робочої
зони у виді 2,4-ДНФГ методом рідинної хроматографії відбір проб повітря
здійснюється на трубці з плівковим сорбентом зі швидкістю 0,5-1,0 л/хв впродовж
5-30 хв. Межа виявлення альдегідів в повітрі 0,015 мг/м3,
погрішність визначення не перевищує 15%. Цей відбір не є селективним, оскільки
в реакцію вступають насичені і ненасичені альдегіди. [3]
Таблиця 2.3 - Хроматографічні методи
визначення акролеїну у формі похідного
Середовище
Реагент
Поглинальне середовище
Чутливість методу
Характеристика методу
Джерело
Атмосферне повітря, кисень працюючої зони
Мета-фенілен-діамін
0,25% р-н гідроксиламіну гідрохлорид,
активоване вугілля
0,07мг/м3 (обʼєм проби 1,5л)
ТШХа
9
Кисень працюючої зони, атмосферне повітря,
повітря в середині приміщення
2,4-дінітрофенілгідразин
Скляні гранули, силіка - гелю,оброблені р-м
2,4-ДНФГ
0,015 мг/м3 (обʼєм проби 30л)
0,05 мг/м3 1,4 мкг/сигарету
ВЕРХб ВЕРХв/УФг
15 2 -(гідроксіметил) пиперідин
Картридж з сорбентом ХАD-2 з насиченим 2
-(гідроксіметил) пиперідином
0,13-1,5 мг/м3 3ppb(обʼєм
проби 48л)
ГХд з азот селективним детектором
29-40
Атмосферне повітря
σ- (2,3,4,5,6-пентафтор
бензил гідроксиламін) (ПФБГА)
0,1 М р-м бісульфіта натрію
5ppb(обʼєм проби 200л)
Капілярна ГХ/МС
41
Атмосферне повітря, повітря в середині
приміщення
дансигідразин
0,29 мкг/дм3
ВЕРХ/ФЛДе з лазерним збудником
35
Примітка: а - тонкошарова
хроматографія, би - високоефективна рідиннахроматографія, в - ультрафіолетовий
детектор, г - газова хроматографія у поєднанні з масс-спектрометрією, д -
газова хроматографія, е - флуориметричний детектор.
У цьому методі вплив, що заважає, на
визначення акролеїну роблять пропіоновий альдегід і ацетон, в присутності яких,
незважаючи на використання градіентного елюювання, не досягається їх повного
розділення. Ця проблема частково розв'язана розробниками фірми Waters, що
запропонували визначати альдегіди і кетон у вигляді 2,4-ДНФГ методом ВЭЖХ на
колонці Nova- Pak C18 3,9*1500 mm. Межа виявлення альдегідів в
повітрі 3 ppb або 0,006 мг/м3. Для уловлювання акролеїну з повітря пропонуються
також дансилгідразин (ДСГ) і 4-гидразинбензойная кислота у картріджах і
пасивних пробовідбірниках (відзначається низька відтворюваність результатів),
пентафлуорофенилгідразин, о-бензил-гидроксиламін, н-бензилэтаноламін, цистамін
і н-метил-4-гидразино-7-нітробензо-фуразан (застосування обмежене у зв'язку з
дорогим устаткуванням для отримання реагентів, недостатньою чутливістю і малою
селективністю).В якості реагенту для уловлювання акролеїну з повітря
застосовується 2 -(гідроксіметил) піперидин. R. із співавторами пропонує для
виявлення акролеїну в повітрі робочої зони метод газової хроматографії у
вигляді похідного оксазолидіну. Проби відбираються зі швидкістю 0,1 дм3/хв..
на сорбент ХАD з нанесеним 2 -(гідроксиметил) піперидином, який при взаємодії з
акролеїном утворює біциклічний оксазолидін. Дериват екстрагують толуолом і
аналізують на газовому хроматографі з азот- селективным детектором на колонці з
Supelcoport +5% SP - 2401 - DB. Авторами експериментально встановлено, що час,
необхідний для завершення реакції акролеїну з 2 -(гідроксиметил) піперидином,
складає 12 годин. Діапазон вимірюваних концентрацій 0,13-1,5 мг/м3 з
погрішністю визначення 11,1%. Відібрані проби стабільні впродовж 28 днів.Y.
Seaman із співавторами пропонує визначати акролеїн в присутностігазоподібних
карбонілівГХметотодомзо (2,3,4,5,6пентафтор- бензил) гідроксил аміном (ПФБГА).
Повітря відбирається 10 хвилин із швидкістю 20 л/хв в скрубер Кофера, що
містить 0,1 М розчин бісульфіту натрію, що підкисляє 0,1 М сірчаною кислотою до
рН 5,0. При взаємодії бісульфіту натрію з акролеїном утворюються сульфонати,
після дисоціації яких карбоніли дериватизують з ПФБГА, що утворює термічно
стабільні адукти. Реакція дериватизації протікає 24 години. Після екстракції
гексаном і висушуванні до об'єму 0,5 см3 пробу аналізують на
газовому хроматографі Agilent з капілярною колонкою DB - XLB і квадрупольним
масс-спектрометром. Чутливість виявлення дорівнює 0,000012 мг/м3, що
дає змогу визначати акролеїн на рівні референтної концентрації. Використання
мас-спектрометрії вирішує проблему селективності і чутливості, разом з тим
складна і тривала обробка пробі (більше 24 годин) недопустима при проведенні
рутинних аналізів. Особливу увагу розробники приділяють відбору проб акролеїну
з повітря. Низький рівень дії і висока реакційна здатність акролеїну є причиною
основних недоліків більшості методів, розроблених для контролю акролеїну в
повітрі. Недоліки такі: обмежений об'єм проби внаслідок проскакування
акролеїну, необхідність зберігання проби при 0 °С, низька міра витягування з
сорбенту, громіздкий пробовідбір, використання токсичних хімікатів і так далі.
Спочатку відбір акролеїну з повітря
проводили в рідкі поглинальні середовища - дистильовану воду, етанол, суміш
перманганату і йодної кислоти, солянокислий спиртовий розчин тіосемікарбазиду,
спиртовий розчин 4-гексилрезорцину, в результаті досягалася невисока міра
збагачення проби. У разі відбору в розчин з дериватизируючим агентом слід
враховувати втрату частини акролеїну, що не вступила в реакцію з реактивом, а
також за рахунок побічних реакцій. Якщо акролеїн слабо утримується поглинаючому
розчині, швидкість аспірації вище за швидкість розчинення або хімічної
взаємодії акролеїну з реагентом, то вірогідне проскакування акролеїну.
В якості твердих сорбентів для
відбору акролеїну використали силікагель, активоване вугілля, діатоміт.
Головною важкістю при концентратуванні на цих сорбентах являється вибір
ефективного способу десорбції, а також накопичення пари води. Полімерні
сорбенти мають ряд переваг в порівнянні з силікагелем і вугіллям, оскільки вони
відносно інертні, гідрофобні і мають велику площу поверхні. Для уловлювання
акролеїну рекомендується використати тенакс ТА, порапак N. Ефективними
адсорбентами для уловлювання легколетучих органічних сполук є молекулярні сита
(цеоліти). Ефект поглинання заснований на особливостях геометричних розмірів
уловлюваних молекул, коли цеоліти поглинають молекулу або частину молекули, що
проходить через мікропори сорбенту в процесі пробовідбору. Для уловлювання
акролеїну з повітря пропонується використати цеоліт 13Х. [9]
Висновки
Дана тема є актуальною. В роботі
були вивчені особливості методу газової хроматографії. Крім класичної
літератури були розглянуті роботі сучасних науковців, що дозволило мені оцінити
актуальність теми та можливість подальшого розвитку цього методу.
Метод газової хроматографії один із
найсучасніших методів аналізу. Його відмінності - експресність, висока
точність, чутливість, можливість автоматизації. Ступінь універсальності і
гнучкості методу газової хроматографії багато чому визначається існуючим
технічним рівнем апаратури.
Були розглянуті практичні методи
застосування газової хроматографії, на підставі цього зроблено такі висновки:
1 На відміну від існуючих
раніше стандартних методів екологічного контролю із застосуванням насадних
колонок, для визначення концентрації парів летких органічних сполук в газових
викидах промислових підприємств методом капілярної газорідинної хроматографії,
були використані макрокапіллярні колонки, висока ефективність яких дозволяє розділити
багатокомпонентну суміш пару летючих органічних сполук без попереднього
концентрування.
2 Проведена методика газового
аналізу на хроматографі "ЦВЕТ-800" з ви користуванням атестованої
газової суміші (Не, H2, Ar, O2, N2, CО2 і
CO). Було встановлено, що в пробах присутні всі газові компоненти, такі, що
заздалегідь передбачалися при виготовленні атестованої суміші, а також метан і
ще один не відомий компонент.
При концентрації акролеїну
з повітря перевага віддається відборам, що забезпечують максимальне уловлювання
аналіту у вигляді його похідного з хімічним реагентом з утворенням нелеткого і
стабільного похідного.
Список використаної літератури
1. Васильев В.П.
Аналитическая химия. В 2ч.Ч.2. Физико-химические методы анализа. / В.П.
Васильев. - М.: Высш. шк., 1989. - 384 с.
. Котов Г.Н., Конопелько
Л.А., Другов Ю.С. Газохроматографическое определение ароматических
углеводородов в городском воздухе// Жур. аналит. хим. 1999. Т.54. Вып 5. С. 531
. Другов Ю.С.,
Ягодовский В.Д. Методы концентрирования при анализе воздуха // В сб.: Успехи
аналитич. химии. - 1990. - Т. 4. - С. 113-143.
4. Дмитриев M.Т., Кознина
Н.И. // Санитарно-химический анализ загрязняющих веществ в окружающей среде.
М.: Химия, 1989. С. 368.
. Столяров Б.Т., Савинов
И.М., Виттенберг А.Г. Руководство к практическим работам по газовой
хроматографии. - Л., Химия, 1988, 223 с.
. Пецев Н., Коцев Н.
Справочник по газовой хроматографии. - М., Мир, 1987, 125 с.
. Анваер Б.И., Другов Ю.
С Газовая хроматография неорганических веществ. - М., Химия, 1976, 31 с.
. Синкувене Д. С.
Гигиеническая оценка акролеина как загрязнителя атмосферного воздуха // Гигиена
и санитария. - 1970.- № 3. - 610 с.
. Уланова Т.С.,
Карнажицкая Т.Д., Пшеничникова Е.О. Аналитический обзор методов определения
микроколичеств акролеина в воздухе. - Издат. дом «АКАДЕМИЯ ЕСТЕСТВОЗНАНИЯ». -
2013. - №5. -с. 456-462.
. Волков и др. /
Сорбционные и хроматографические процессы -2009, т. 9, вып. 6, - с. 862-866.
. Мустафин Р.Н.,
Абдигамитова А.А., Дерявко И.И. Возможности определения состава сложной газовой
смеси. - «Научно-технический журнал», 2006, вып. 1, -с. 17-21.
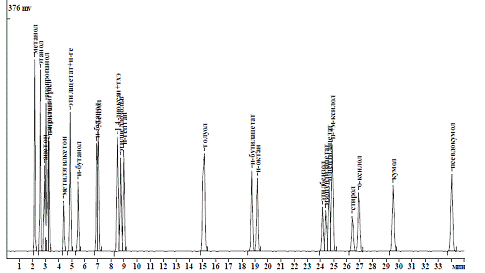
+ 50 °С, витрата 20 см3/хв..), після третього - тільки СО2
(колонка з полісорбом, температура + 50 °С, витрата 30 см3/хв..).
Час утримування компонентів суміші на сорбентах, отримані за таких режимів,
склали для H2, Ar, O2, N2, CO2 і CO
відповідно до 50, 192, 220, 1090, 1260 і 1551, а загальна тривалість
хроматографування суміші виявилася менше 28 хв.