Материал: mgk_voprosy_dlya_samopodgotovki
Вопросы для самоподготовки:
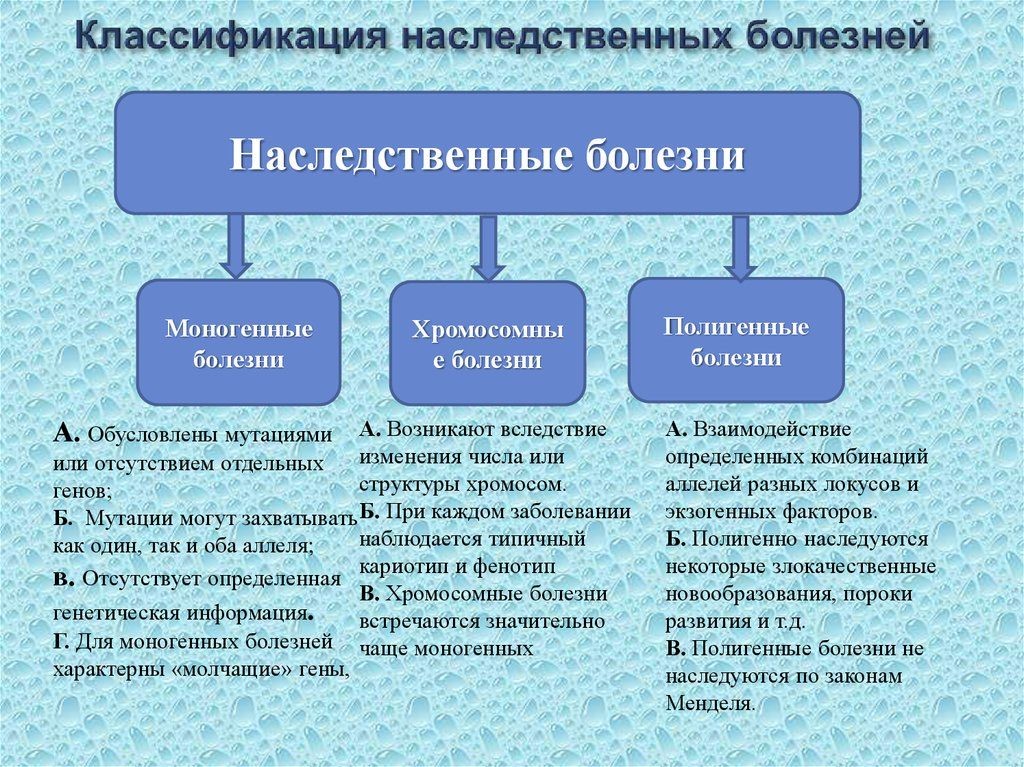
1. Классификации наследственных болезней.
2.Характеристика (генотипическая и фенотипическая) основных хромосомных синдромов (аутосомных: Дауна, Патау, Эдвардса, трисомии хромосомы 8, «кошачьего крика», Вольфа-Хиршхорна).
Классификация моногенных болезней.
Характеристика наиболее распространённых наследственных болезней обмена веществ (фенилкетонурия, алкаптонурия, галактоземия, муковисцидоз).
5.Мультифакториальные болезни (с наследственной предрасположенностью). Характеристика генетических и внешнесредовых факторов, являющихся причинами появления болезней. Понятие «порога подверженности».
6. Понятие и классификация врожденных пороков развития.
7.Наследственные болезни с нетрадиционным наследованием: митохондриальные болезни, болезни импринтинга (синдром Прадера-Вилли, Энгельмана); экспансии тринуклеотидных повторов (синдром ломкой Х-хромосомы Мартина-Белла). Генотипическая и фенотипическая характеристика.
8.Принципы профилактики наследственных болезней.
9.Медико-генетическое консультирование как учреждение и врачебное заключение. Виды, этапы проведения, задачи.
10.Пренатальная диагностикиа. Методы (неинвазивные и инвазивные): УЗИ, амниоцентез, биопсия хориона, кордоцентез, фетоскопия, определение маркёров: альфа-фетопротеина, хорионического гонадотропина и др.). Показания к проведению, значение.
1
Вопросы для самоподготовки:
1. Классификации наследственных болезней.
2.Характеристика (генотипическая и фенотипическая) основных хромосомных синдромов (аутосомных: Дауна, Патау, Эдвардса, трисомии хромосомы 8, «кошачьего крика», Вольфа-Хиршхорна).
Хромосомные синдромы. Аутосомные синдромы.
1.Синдром Дауна.
Клиническое описание синдрома сделано в 1866 году. Частота 1: 550 – 700 новорождённых среди умственно отсталых детей выявляется 10-12 % больных синдромом Дауна. Выделяют 3 формы:
простая трисомия по хромосоме 21. Встречаются в 95% всех случаев.
транслокация 21 хромосомы на другие ( чаще на 15, реже на 14, ещё реже на 21, 22, У- хромосому) – 4% случаев.
мозаичный вариант синдрома – 1%
Фенотипы: брахицефалическая форма черепа с ускорением переднее–заднего размера и утолщением затылка, избыток кожи на затылке, плоский профиль лица, эпикант (вертикальная кожная складка у внутреннего угла глазной щели), монголоидный разрез глазных щелей, помутнение хрусталика, косоглазие, короткий нос с широким плоским переносьем, полуоткрытый рот с толстыми губами и высунутым языком (макроглоссия), узкое и короткое нёбо. Руки короткие и широкие, поперечная складка ладоней. Часто наблюдаются врождённые пороки сердца (дефект межжелудочковой перегородки, открытый артериальный проток), деформация скелета. Умственная отсталость (имбицильность, дебильность.)
2.Синдром Патау
Частота встречаемости 1: 6000 новорождённых. Различают три формы:
простая трисомия по хромосоме 13 (75 % случаев); - транслокация (чаще робертсоновская) (20 % случаев);
мозаичная (5%).
Фенотипы: микроцефалия, тригоноцефалия (расширение черепа в затылочной и сужение в лобной части), узкие глазные щели, широкое основание носа, низко посаженные деформированные уши, микрофтальмия (малые размеры глазного яблока), микрогнатия (малые размеры верхней челюсти), расщелина губы и нёба, полидактилия, пороки внутренних органов (головного мозга, сердца и сосудов, почек, пищеварения, половых органов). Дети погибают обычно в течение первых трёх месяцев жизни.
3.Синдром Эдвардса
Частота встречаемости 1: 7000. Различают 2 формы:
трисомная по хромосоме 18 (90 % случаев);
мозаичная (10 % случаев).
Фенотип: задержка роста, множественные аномалии развития: долихоцефалический череп (преобладание продольных размеров головы над поперечным) с выступающим затылком, «птичий» профиль лица, короткие и горизонтально расположенные глазные щели; маленькие, деформированные низко расположенные уши, избыточная кожа на затылке; микростомия (узкая ротовая щель); флексорное сгибание кисти с наложением указательного пальца на III, а V на IV; «стопа – качалка» (с провисающим сводом и выступающей кзади пяткой); синдактилия; врождённые пороки сердца и крупных сосудов. Продолжительность жизни резко снижена.
4.Трисомия по хромосоме 8 .
Частота встречаемости 1: 50000. Различают 2 формы:
мозаичный вариант (84 % случаев);
трисомия по хромосоме 8 (16 % случаев).
+
Фенотип: множественные изменения опорно–двигательного аппарата; туловище длинное, слияние рёбер и позвонков, сколиоз, кифоз , spina bifida в грудном или в поясничном отделах (расщепление позвоночного столба); ограничены движения в суставах. Отмечается выраженный черепо-лицевой дисморфизм: большой квадратный череп, выступающий лоб, гипертелоризм, косоглазие,
микрогнатия, вывернутая нижняя губа, короткая шея, низко расположенные деформированные уши. Пороки внутренних органов: подковообразная почка, гидронефроз, гипоплазия гениталий. Задержка физического и интеллектуального развития.
3. Классификация моногенных болезней.
Классификация:
1. По типу наследования:
МЗ с аутосомно-доминантным типом наследования: хорея Гентингтона, нейрофиброматоз, синдромы Марфана, Крузона, Холт-Орама, ПМД
лицелопаточноплечевая, оливопонтоцеребеллярная атрофия I типа, ахондроплазия и др.;
МЗ с аутосомно-рецессивным типом наследования: большинство наследственных болезней обмена веществ (фенилкетонурия, галактоземия, мукополисахаридозы и др.), атаксия Фридрейха, спинальная мышечная атрофия I-III типов, альбинизм и др.;
МЗ с Х-сцепленным рецессивным наследованием: ПМД Дюшенна/Беккера, ПМД
Эмери-Дрейфуса, гемофилия А и В, бульбоспинальная амиотрофия Кеннеди и др;
МЗ с Х-сцепленным доминантным наследованием: невральная амиотрофия
Шарко-Мари-Тус, витамин-Д резистентный рахит, синдром недержания пигмента и др.;
МЗ с митохондриальным (материнским) наследованием: синдромы MELAS, Кернс-Сейра, Пирсона, MERRF и др.
2. В зависимости от того, какая система организма поражается в большей степени:
болезни нервной системы: нервно-мышечные, с преимущественным поражением пирамидной, экстрапирамидной, координаторной систем, лейкодистрофии и др.;
болезни сердечно-сосудистой системы: семейные формы гиперхолестеринемии, гиперлипопротеидемии, сердечный гликогеноз,
наследственный амилоидоз и др.;
болезни дыхательной системы: идиопатический диффузный фиброз легких, наследственный спонтанный пневмоторакс, изолированный легоч- ный гемосидероз и др.;
болезни желудочно-кишечного тракта: синдром мальабсорбции, врожденная хлоридная диарея, врожденный дефицит лактазы, целиакия и
др.;
болезни соединительной ткани и скелета: несовершенный остеогенез, ахондроплазия, различные формы хондродистрофии, синдром
Элерса-Данлоса, мукополисахаридозы и др.;
болезни кожи и ее придатков: ладонно-подошвенная кератодермия, синдром недержания пигмента, пигментная ксеродерма, ихтиоз, синдром Блума и др.;
болезни почек и мочевыводящих путей: наследственный нефрит, поликистоз почек, несахарный диабет, почечный канальцевый ацидоз и др.;
болезни эндокринных органов: врожденный гипотиреоз, гипофизарный нанизм, гипертиреоз и др.;
болезни органов зрения: анофтальмия врожденная,
болезни органов слуха: рахличные типы наследственной глухоты, синдром микротии с атрезией наружного слухового прохода и др.; - болезни половой системы и др.
По этиологии:
Болезни с установленным первичным генетическим дефектом (т.е в зависимости от типа мутации). Этот принцип наиболее сложен, т.к. одно и то же заболевание может быть обусловлено сотнями различных мутаций (для муковисцидоза установлено около 1000 вариантов мутаций), однако он наиболее точен. В настоящее время доля заболеваний с установленным генетическим дефектом невелика, однако она постоянно растет (можно предположить, что у человека минимально может быть до 30-50 тыс. моногенных заболеваний).
Болезни с неустановленным первичным генетическим дефектом, т.е. около 90% известных на сегодняшний момент форм МЗ.
Болезни с установленным первичным биохимическим дефектом, т.е в зависимости от того, функция какого субстрата нарушена (белок, фермент и др.). На эту группу МЗ, как и в предыдущем случае приходится меньшая часть заболеваний – около 10%.
Болезни с неустановленным первичным биохимическим дефектом. В ближайшее десятилетие предполагается изучить первичные биохимические дефекты основной группы наследственных болезней обмена (НБО).
Характеристика наиболее распространённых наследственных болезней обмена веществ (фенилкетонурия, алкаптонурия, галактоземия, муковисцидоз).
Гипотиреоз. Патология щитовидной железы, которая может привести к отставанию в физическом и психическом развитии. На сегодняшний день своевременно диагностированный гипотиреоз хорошо поддается гормональной терапии.
Распространенность заболевания — 1 случай из 5 тысяч. Андрогенитальный синдром. Патология коры надпочечников, при которой нарушается нормальная выработка гормона кортизола. Может проявиться в виде задержки развития половой системы, проблем с сосудами и сердцем. Полному излечению этот синдром не поддается, но его можно держать под контролем при помощи гормональной терапии. Распространенность заболевания — 1 случай из 15 тысяч.
Муковисцидоз. Заболевание проявляется заметным сгущением секрета в пищеварительном тракте и легких, что приводит к поражениям печени, ЖКТ, дыхательной системы и других органов. Поддается лечению. Распространенность заболевания — 1 случай из 3 тысяч.
Фенилкетонурия. Заболевание, которое характеризуется нарушением выработки определенных ферментов. Последствия достаточно тяжелые. В первую очередь к ним относятся поражения ЦНС. Однако их можно избежать при помощи специальной диеты. Распространенность заболевания — 1 случай из 15 тысяч.
+
Галактоземия. Так называют недостаток фермента, расщепляющего галактозу — один из сахаров, который содержится в лактозе и иных веществах. Последствия нехватки этого
фермента проявляются через несколько недель жизни. У ребенка начинается желтуха, рвота, потеря аппетита. Со временем развиваются тяжелые патологии печени,
замедляется умственное и физическое развитие, ухудшается зрение. Эта врожденная патология опасна, при этом встречается достаточно редко. Распространенность — 1 случай из 30 тысяч.
Алкаптонурия

Алкаптонурия – генетически обусловленное нарушение метаболизма,
характеризующееся врожденным дефицитом фермента гомогентизиназы и приводящее к неполному расщеплению гомогентизиновой кислоты, ее экскреции с мочой и отложению данного метаболита в тканях (коже, суставных хрящах, сухожилиях, склерах и др.). Признаки алкаптонурии появляются в детстве и включают выделение быстро темнеющей на воздухе мочи, пигментацию кожи и склер, остеоартроз, нефролитиаз, охриплость голоса, дисфагию и т. д. Диагноз алкаптонурии устанавливается с учетом клинических проявлений и лабораторных тестов (биохимического исследования мочи, анализа синовиальной жидкости), рентгенографии, артроскопии. Лечение алкаптонурии носит симптоматический характер (диетотерапия, витамин С, хондропротекторы, НПВС, бальнеофизиотерапия); при необходимости проводится протезирование суставов или клапанов сердца.
Общие сведения
Алкаптонурия (гомогентизиновая ацидурия, наследственный охроноз) – наследственная патология обмена веществ, в основе которой лежит нарушение метаболизма тирозина, приводящее к избыточному образованию промежуточного метаболита - гомогентизиновой кислоты. Аккумуляция в организме гомогентизиновой кислоты и ее выделение с мочой и определяют течение симптомокомплекса, известного как алкаптонурия. Поскольку заболевание сопровождается полисистемным поражением, оно представляет
практический интерес для ряда медицинских дисциплин: ревматологии, дерматологии, ортопедии, урологии, кардиохирургии, генетики и др. Алкаптонурия встречается редко, с частотой 1:250 тыс.-1:1 млн. людей в мире. В большей степени патология распространена среди мужского населения Доминиканской республики, Германии, Чехии, Словакии, Индии, США.
Причины алкаптонурии
Алкаптонурия относится к числу генетически обусловленных энзимопатий, наследуемых по аутосомно-рецессивному типу, т. е. для развития клинических проявлений заболевания ребенок должен получить от каждого из родителей по одной копии мутантного гена. При алкаптонурии мутации затрагивают ген оксидазы гомогентизиновой кислоты (HGD), локализованный на длинном плече 3 хромосомы (3q 21-23). В результате нарушается воспроизводство фермента гомогентизиназы (гомогентизиновой оксидазы), который участвует в расщеплении тирозина и фенилаланина.
В норме гомогентизиновая кислота, являющаяся промежуточным метаболитом обмена тирозина и фенилаланина, под действием гомогентизиназы трансформируется в малеилацетоуксусную кислоту и далее расщепляется до ацетоуксусной и фумаровой кислот, которые затем участвуют в последующих биохимических циклах. При алкаптонурии, в условиях врожденного дефицита фермента, гомогентизиновая кислота не подвергается дальнейшему метаболизму, а превращается в хиноновые полифенолы (пигмент алкаптон), которые накапливаются в соединительных тканях и в больших количествах экскретируются с мочой (до 4-8 г в сутки).
Симптомы алкаптонурии
Алкаптонурия характеризуется следующими основными симптомокомплексами:
гомогентизиновой ацидурией, охронозом и артропатией. Эти признаки возникают в разное время: окрашивание мочи существует с рождения, пигментация тканей становится выраженной к 30 годам, поражение суставов развивается на четвертом десятилетии жизни.
Ранние признаки алкаптонурии можно заметить еще в раннем детском возрасте: на мокрых пеленках ребенка остаются темные разводы от мочи, которые невозможно отстирать. Из-за большого количества гомогентизиновой кислоты собранная моча при отстаивании также быстро приобретает темно-бурый цвет. В дальнейшем со стороны мочеполовых органов нередко развиваются пиелонефрит, мочекаменная болезнь, калькулезный простатит.
Кожный синдром при алкаптонурии характеризуется появлением серо-коричневой пигментации на лице (в области спинки носа, вокруг губ и глаз), на шее, ладонях, животе, подмышечной и паховой области. Типичным признаком алкаптонурии служит уплотнение и серо-голубое окрашивание ушных раковин, пигментация склеры и конъюнктивы. Диффузное отложение пигмента отмечается в хрящах гортани, что сопровождается охриплостью голоса, одышкой, дисфагией и болью при глотании. Со временем развивается кальцификация аорты и клапанов сердца, следствием чего являются атеросклероз, приобретенные аортальный и митральный пороки. При тяжелых формах алкаптонурии отложение пигмента может встречаться в щитовидной железе, надпочечниках, яичках, поджелудочной железе, селезенке.
Поражение опорно-двигательной системы при алкаптонурии, затрагивает, главным образом, крупные суставы нижних конечностей и позвоночник. Раньше всего изменения по типу деформирующего спондилеза развиваются в поясничном, затем в грудном отделе позвоночника. При этом постепенно нарастает сглаженность поясничного лордоза, боли и скованность при движениях, вплоть до полной потери подвижности в пораженных отделах позвоночника. Вслед за поражением позвоночника появляются признаки деформирующего остеоартроза коленных, плечевых и тазобедренных суставов. Характерны артралгии, припухлость суставов (реактивный синовит), крепитация, подвижность, развитие сгибательных контрактур. Нередко при алкаптонурии поражаются крестцово-подвздошные суставы и лонное сочленение.
Галактоземия - наследственная ферментопатия, характеризующаяся нарушением нормального процесса углеводного обмена, а именно – метаболизма галактозы. Признаками галактоземии являются непереносимость грудного молока и молочных смесей, рвота, анорексия, гипотрофия, желтуха, цирроз печени, спленомегалия, отеки, катаракта, задержка психомоторного развития. Скрининг на галактоземию проводится всем новорожденным; дополнительное обследование включает определение уровня галактозы в крови и моче, проведение нагрузочных проб с галактозой и глюкозой, генетическое тестирование, УЗИ брюшной полости, ЭЭГ и др. Основу терапии галактоземии составляет безлактозная диета, назначаемая с первых дней жизни ребенка.